Genetics Department, University Medical Center Groningen, Groningen, the Netherlands.
Genomics Coordination Center, University of Groningen, University Medical Center Groningen, Groningen, the Netherlands.
Genome Biol. 2023 Apr 18;24(1):80. doi: 10.1186/s13059-023-02897-x.
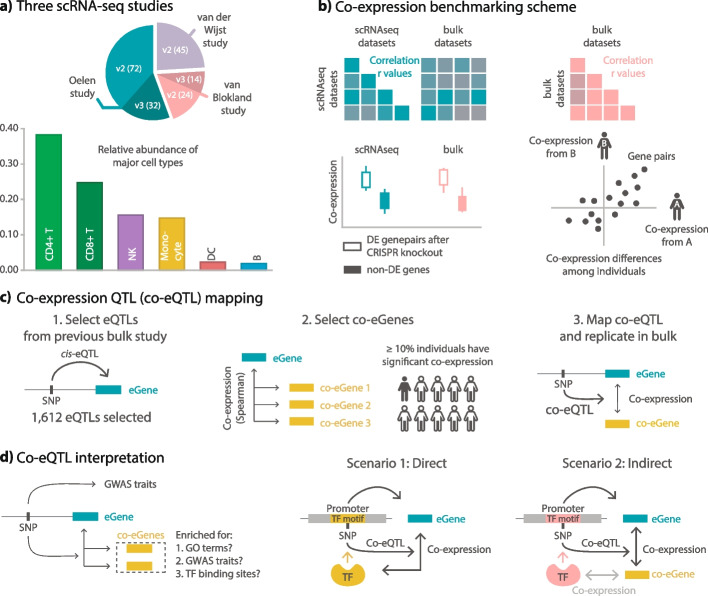
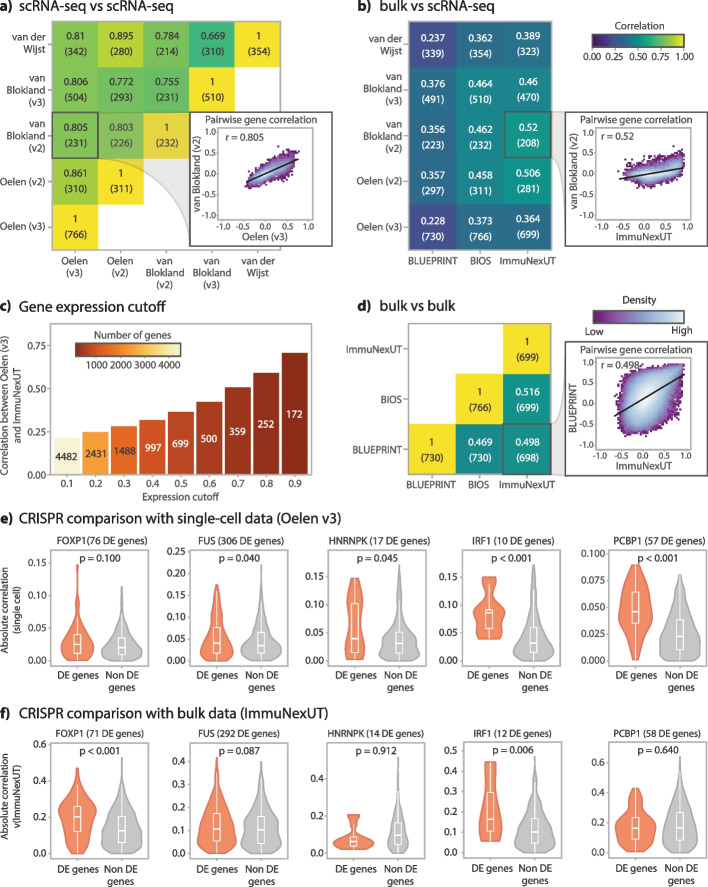
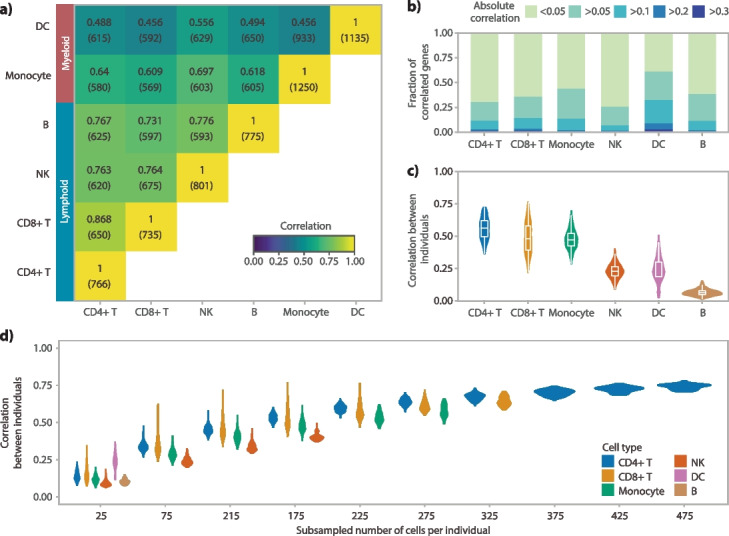
Expression quantitative trait loci (eQTL) studies show how genetic variants affect downstream gene expression. Single-cell data allows reconstruction of personalized co-expression networks and therefore the identification of SNPs altering co-expression patterns (co-expression QTLs, co-eQTLs) and the affected upstream regulatory processes using a limited number of individuals.
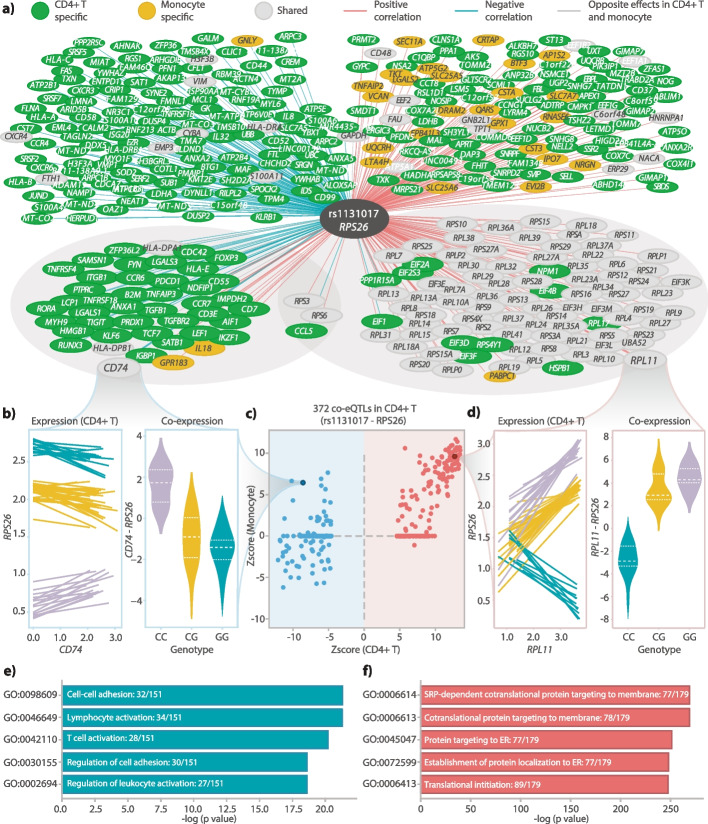
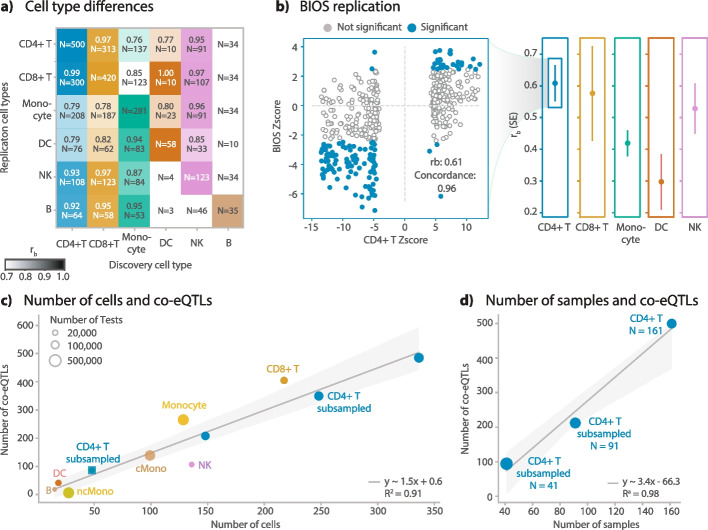
We conduct a co-eQTL meta-analysis across four scRNA-seq peripheral blood mononuclear cell datasets using a novel filtering strategy followed by a permutation-based multiple testing approach. Before the analysis, we evaluate the co-expression patterns required for co-eQTL identification using different external resources. We identify a robust set of cell-type-specific co-eQTLs for 72 independent SNPs affecting 946 gene pairs. These co-eQTLs are replicated in a large bulk cohort and provide novel insights into how disease-associated variants alter regulatory networks. One co-eQTL SNP, rs1131017, that is associated with several autoimmune diseases, affects the co-expression of RPS26 with other ribosomal genes. Interestingly, specifically in T cells, the SNP additionally affects co-expression of RPS26 and a group of genes associated with T cell activation and autoimmune disease. Among these genes, we identify enrichment for targets of five T-cell-activation-related transcription factors whose binding sites harbor rs1131017. This reveals a previously overlooked process and pinpoints potential regulators that could explain the association of rs1131017 with autoimmune diseases.
Our co-eQTL results highlight the importance of studying context-specific gene regulation to understand the biological implications of genetic variation. With the expected growth of sc-eQTL datasets, our strategy and technical guidelines will facilitate future co-eQTL identification, further elucidating unknown disease mechanisms.
表达数量性状基因座(eQTL)研究表明遗传变异如何影响下游基因表达。单细胞数据允许重建个性化的共表达网络,因此可以使用少数个体识别改变共表达模式的 SNP(共表达 QTL,co-eQTL)和受影响的上游调节过程。
我们使用一种新的过滤策略和基于置换的多重测试方法,对四个 scRNA-seq 外周血单核细胞数据集进行了 co-eQTL 元分析。在分析之前,我们使用不同的外部资源评估了识别 co-eQTL 所需的共表达模式。我们确定了一组稳健的细胞类型特异性 co-eQTL,用于影响 946 个基因对的 72 个独立 SNP。这些 co-eQTL 在一个大型批量队列中得到了复制,并为疾病相关变异如何改变调节网络提供了新的见解。一个与多种自身免疫性疾病相关的 co-eQTL SNP rs1131017 影响了 RPS26 与其他核糖体基因的共表达。有趣的是,特别是在 T 细胞中,该 SNP 还影响了 RPS26 与一组与 T 细胞激活和自身免疫性疾病相关的基因的共表达。在这些基因中,我们发现了与五个与 T 细胞激活相关的转录因子的靶基因富集,其结合位点含有 rs1131017。这揭示了一个以前被忽视的过程,并指出了潜在的调节剂,这些调节剂可以解释 rs1131017 与自身免疫性疾病的关联。
我们的 co-eQTL 结果强调了研究特定于上下文的基因调控以了解遗传变异的生物学意义的重要性。随着 sc-eQTL 数据集的预期增长,我们的策略和技术指南将促进未来 co-eQTL 的识别,进一步阐明未知的疾病机制。