Department of Epidemiology and Biostatistics, School of Public Health, Anhui Medical University, Hefei, Anhui, China
Teaching Center for Preventive Medicine, School of Public Health, Anhui Medical University, Hefei, China.
RMD Open. 2022 Sep;8(2). doi: 10.1136/rmdopen-2022-002529.
Although genome-wide association studies (GWASs) have identified more than 100 loci associated with rheumatoid arthritis (RA) susceptibility, the causal genes and biological mechanisms remain largely unknown.
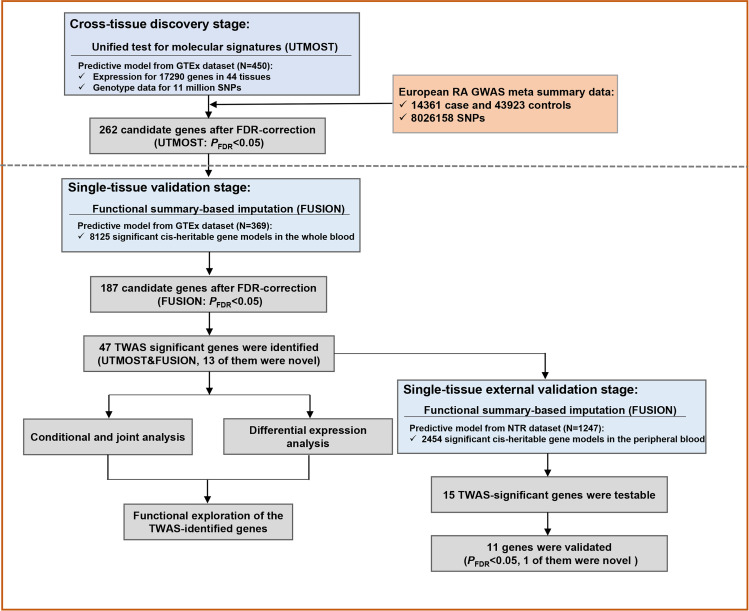
A cross-tissue transcriptome-wide association study (TWAS) using the unified test for molecular signaturestool was performed to integrate GWAS summary statistics from 58 284 individuals (14 361 RA cases and 43 923 controls) with gene-expression matrix in the Genotype-Tissue Expression project. Subsequently, a single tissue by using FUSION software was conducted to validate the significant associations. We also compared the TWAS with different gene-based methodologies, including Summary Data Based Mendelian Randomization (SMR) and Multimarker Analysis of Genomic Annotation (MAGMA). Further in silico analyses (conditional and joint analysis, differential expression analysis and gene-set enrichment analysis) were used to deepen our understanding of genetic architecture and comorbidity aetiology of RA.
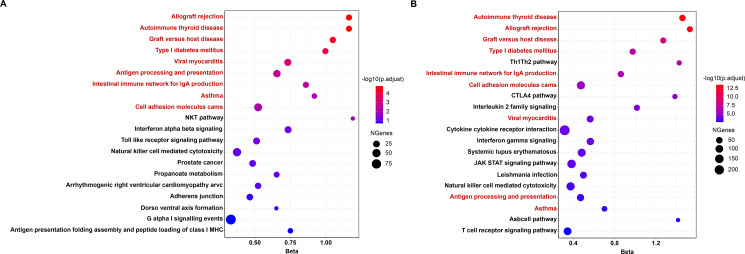
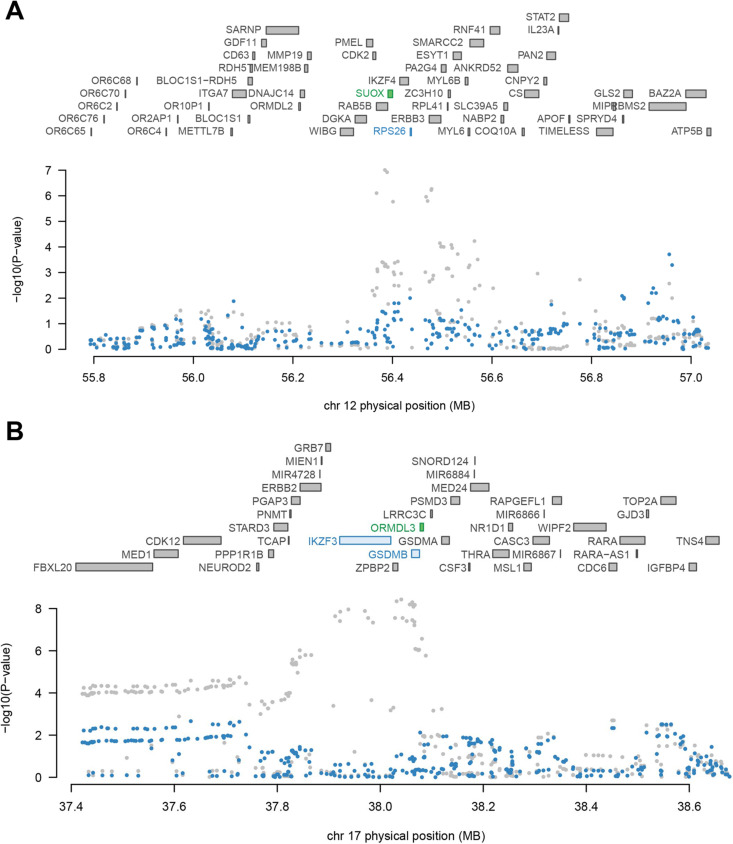
We identified a total of 47 significant candidate genes for RA in both cross-tissue and single-tissue test after multiple testing correction, of which 40 TWAS-identified genes were verified by SMR or MAGMA. Among them, 13 genes were situated outside of previously reported significant loci by RA GWAS. Both TWAS-based and MAGMA-based enrichment analyses illustrated the shared genetic determinants among autoimmune thyroid disease, asthma, type I diabetes mellitus and RA.
Our study unveils 13 new candidate genes whose predicted expression is associated with risk of RA, providing new insights into the underlying genetic architecture of RA.
尽管全基因组关联研究(GWAS)已经确定了 100 多个与类风湿关节炎(RA)易感性相关的基因座,但这些基因的因果关系和生物学机制在很大程度上仍然未知。
使用分子特征统一测试工具进行跨组织转录组全基因组关联研究(TWAS),将来自 58284 个人的 GWAS 汇总统计数据(包括 14361 例 RA 病例和 43923 例对照)与基因型组织表达计划中的基因表达矩阵进行整合。随后,使用 FUSION 软件进行单组织验证,以验证显著关联。我们还将 TWAS 与不同的基于基因的方法进行了比较,包括基于汇总数据的孟德尔随机化(SMR)和基因组注释的多标记分析(MAGMA)。进一步的计算分析(条件和联合分析、差异表达分析和基因集富集分析)用于深入了解 RA 的遗传结构和共病病因。
在经过多次测试校正后,我们在跨组织和单组织测试中总共确定了 47 个与 RA 相关的显著候选基因,其中 40 个 TWAS 鉴定的基因被 SMR 或 MAGMA 验证。在这些基因中,有 13 个基因位于 RA GWAS 先前报道的显著基因座之外。基于 TWAS 的和基于 MAGMA 的富集分析都说明了自身免疫性甲状腺疾病、哮喘、I 型糖尿病和 RA 之间存在共同的遗传决定因素。
我们的研究揭示了 13 个新的候选基因,它们的预测表达与 RA 的发病风险相关,为 RA 的潜在遗传结构提供了新的见解。