Shenzhen People's Hospital, First Affiliated Hospital of Southern University of Science and Technology, Second Clinical Medicine College of Jinan University, Shenzhen 518020, China.
Department of Computer Science and Engineering, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong.
Bioinformatics. 2023 May 4;39(5). doi: 10.1093/bioinformatics/btad213.
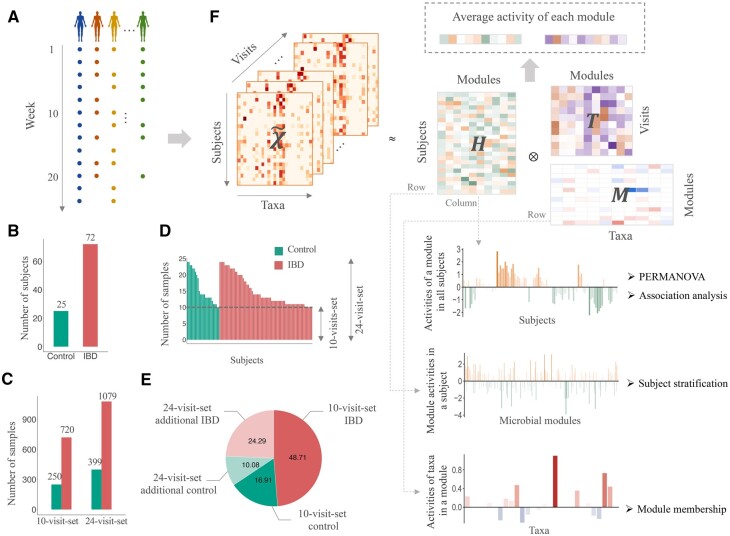
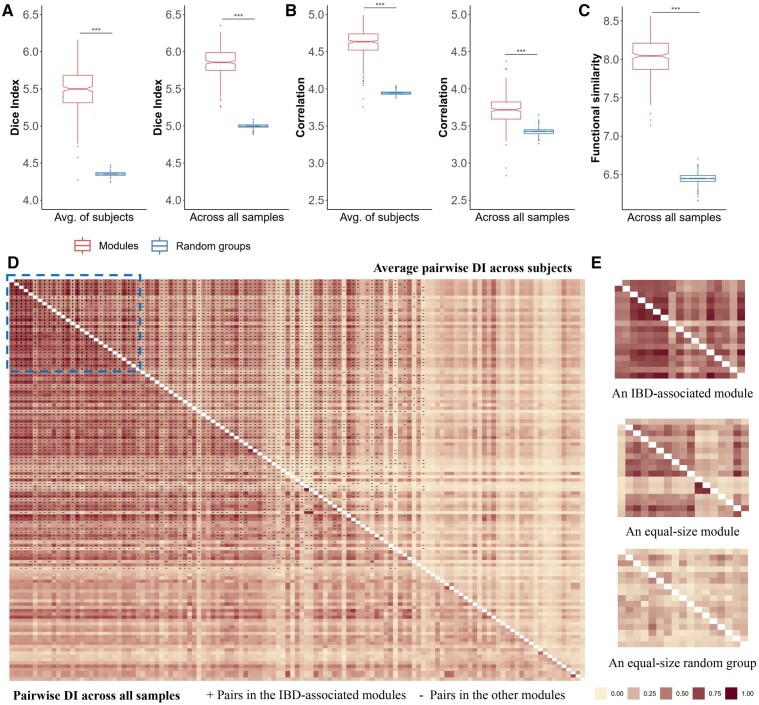
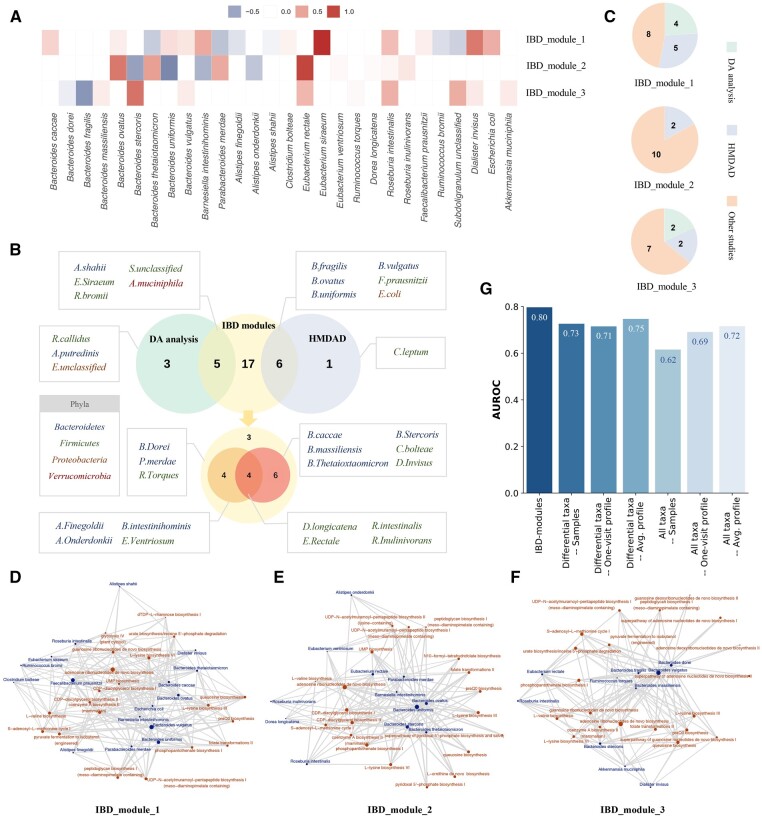
Human gut microbiota plays a vital role in maintaining body health. The dysbiosis of gut microbiota is associated with a variety of diseases. It is critical to uncover the associations between gut microbiota and disease states as well as other intrinsic or environmental factors. However, inferring alterations of individual microbial taxa based on relative abundance data likely leads to false associations and conflicting discoveries in different studies. Moreover, the effects of underlying factors and microbe-microbe interactions could lead to the alteration of larger sets of taxa. It might be more robust to investigate gut microbiota using groups of related taxa instead of the composition of individual taxa.
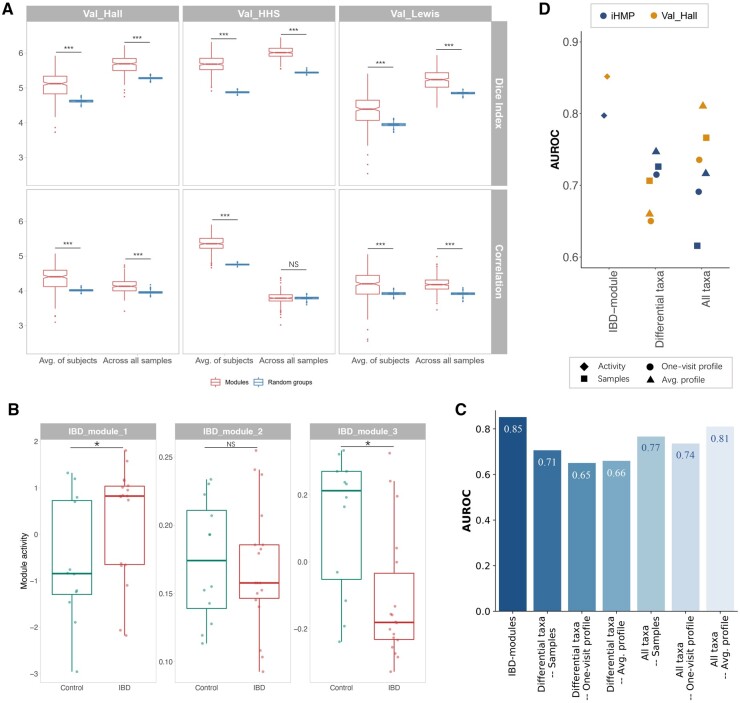
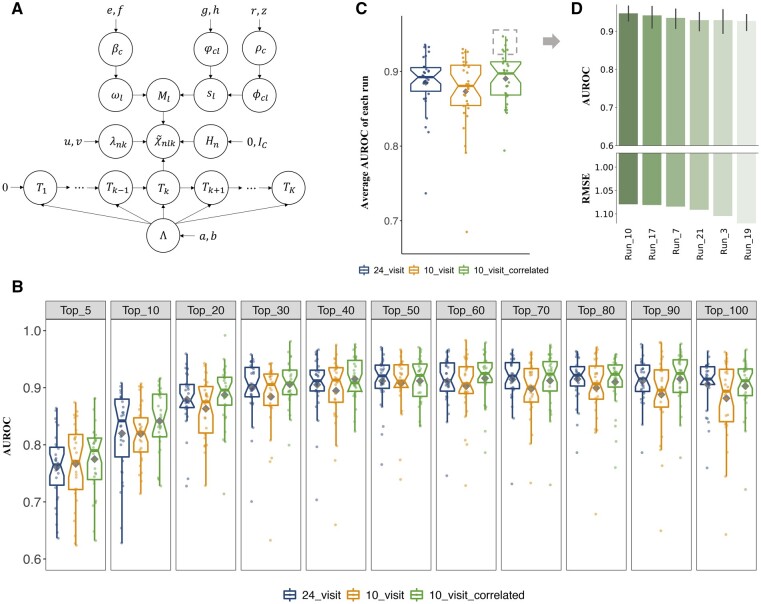
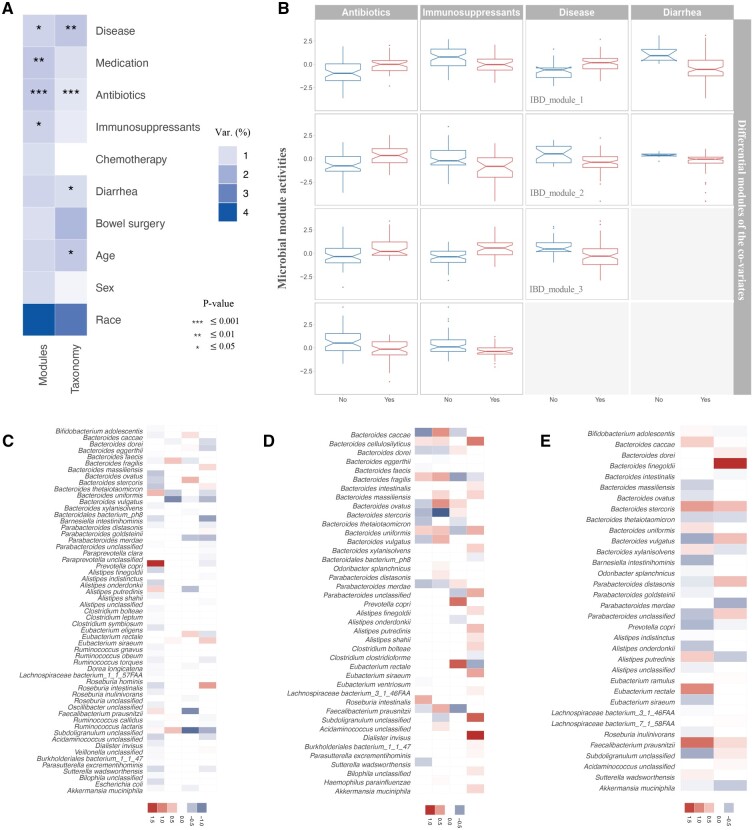
We proposed a novel method to identify underlying microbial modules, i.e. groups of taxa with similar abundance patterns affected by a common latent factor, from longitudinal gut microbiota and applied it to inflammatory bowel disease (IBD). The identified modules demonstrated closer intragroup relationships, indicating potential microbe-microbe interactions and influences of underlying factors. Associations between the modules and several clinical factors were investigated, especially disease states. The IBD-associated modules performed better in stratifying the subjects compared with the relative abundance of individual taxa. The modules were further validated in external cohorts, demonstrating the efficacy of the proposed method in identifying general and robust microbial modules. The study reveals the benefit of considering the ecological effects in gut microbiota analysis and the great promise of linking clinical factors with underlying microbial modules.
人类肠道微生物群在维持身体健康方面起着至关重要的作用。肠道微生物群的失调与多种疾病有关。揭示肠道微生物群与疾病状态以及其他内在或环境因素之间的关联至关重要。然而,根据相对丰度数据推断个体微生物分类群的变化可能导致不同研究中出现虚假关联和相互矛盾的发现。此外,潜在因素和微生物-微生物相互作用的影响可能导致更大的分类群发生改变。使用相关分类群组而不是单个分类群的组成来研究肠道微生物群可能更为稳健。
我们提出了一种从纵向肠道微生物群中识别潜在微生物模块的新方法,即受共同潜在因素影响的具有相似丰度模式的一组分类群,并将其应用于炎症性肠病(IBD)。所鉴定的模块表现出更紧密的组内关系,表明潜在的微生物-微生物相互作用和潜在因素的影响。研究了这些模块与几种临床因素之间的关联,特别是疾病状态。与个体分类群的相对丰度相比,IBD 相关模块在对受试者进行分层方面表现更好。该模块在外部队列中进一步得到验证,证明了所提出的方法在识别一般和稳健的微生物模块方面的有效性。该研究揭示了在肠道微生物群分析中考虑生态效应的益处,以及将临床因素与潜在微生物模块联系起来的巨大前景。