Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA.
Institute for Medical Biometry and Bioinformatics, Medical Faculty, Heinrich Heine University, 40225 Düsseldorf, Germany.
Genome Res. 2023 Apr;33(4):496-510. doi: 10.1101/gr.277334.122. Epub 2023 May 10.
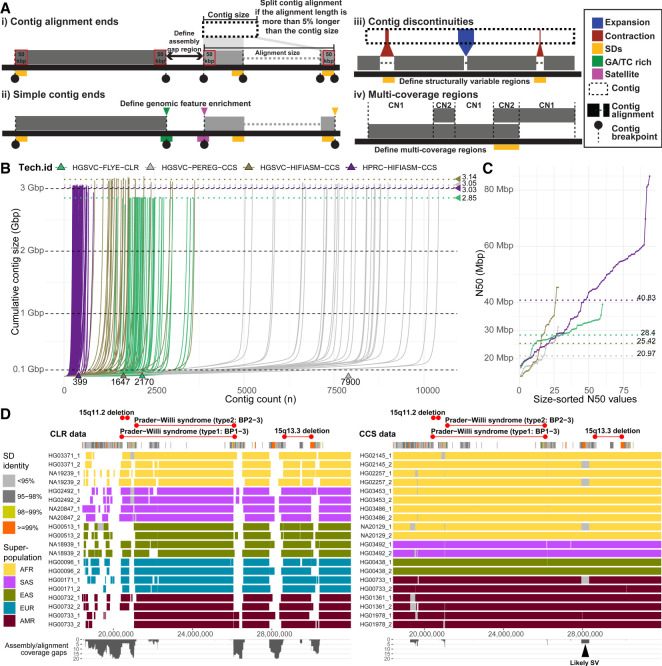
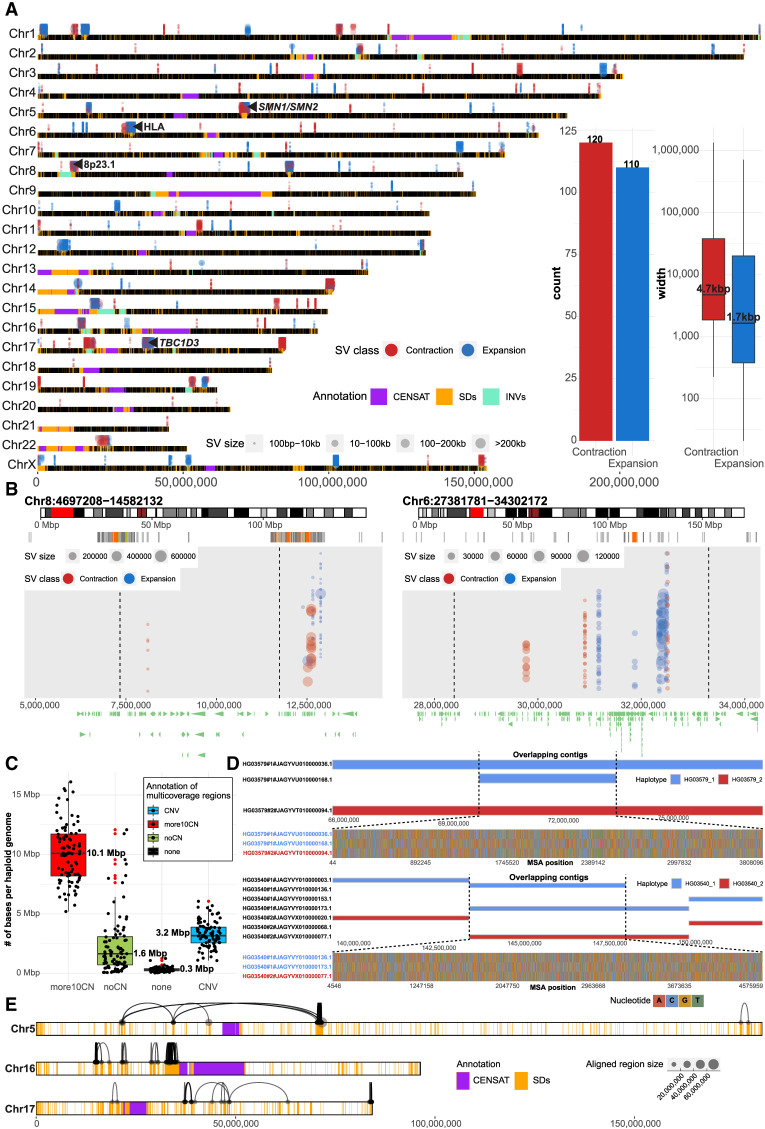
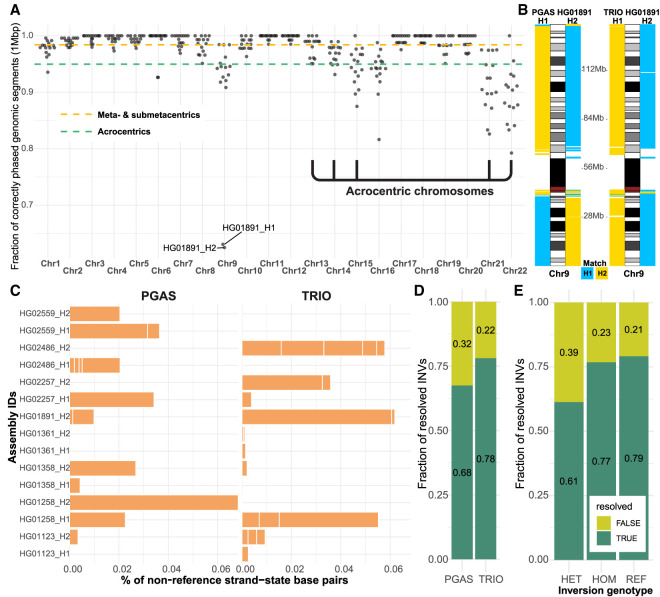
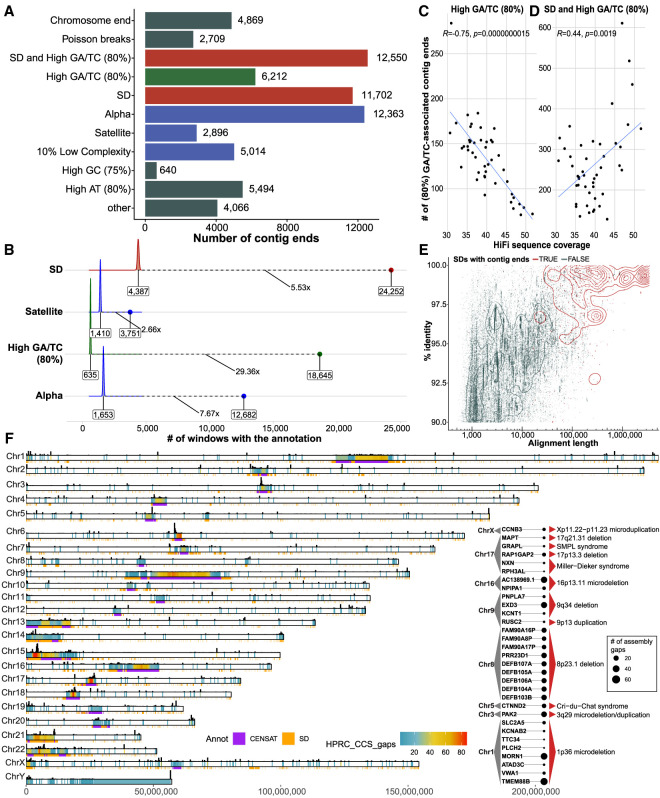
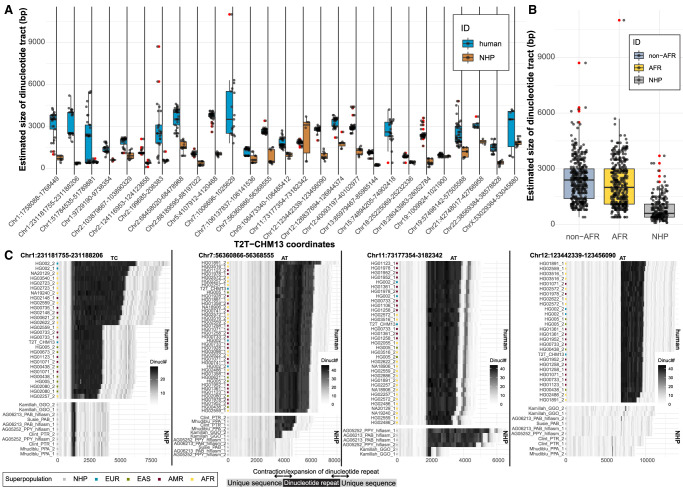
There has been tremendous progress in phased genome assembly production by combining long-read data with parental information or linked-read data. Nevertheless, a typical phased genome assembly generated by trio-hifiasm still generates more than 140 gaps. We perform a detailed analysis of gaps, assembly breaks, and misorientations from 182 haploid assemblies obtained from a diversity panel of 77 unique human samples. Although trio-based approaches using HiFi are the current gold standard, chromosome-wide phasing accuracy is comparable when using Strand-seq instead of parental data. Importantly, the majority of assembly gaps cluster near the largest and most identical repeats (including segmental duplications [35.4%], satellite DNA [22.3%], or regions enriched in GA/AT-rich DNA [27.4%]). Consequently, 1513 protein-coding genes overlap assembly gaps in at least one haplotype, and 231 are recurrently disrupted or missing from five or more haplotypes. Furthermore, we estimate that 6-7 Mbp of DNA are misorientated per haplotype irrespective of whether trio-free or trio-based approaches are used. Of these misorientations, 81% correspond to bona fide large inversion polymorphisms in the human species, most of which are flanked by large segmental duplications. We also identify large-scale alignment discontinuities consistent with 11.9 Mbp of deletions and 161.4 Mbp of insertions per haploid genome. Although 99% of this variation corresponds to satellite DNA, we identify 230 regions of euchromatic DNA with frequent expansions and contractions, nearly half of which overlap with 197 protein-coding genes. Such variable and incompletely assembled regions are important targets for future algorithmic development and pangenome representation.
通过将长读数据与亲本信息或链接读数据相结合,在分阶段基因组组装生产方面取得了巨大进展。然而,通过 trio-hifiasm 生成的典型相位基因组仍然会产生超过 140 个缺口。我们对来自 77 个独特人类样本多样性面板的 182 个单体组装获得的缺口、组装断裂和定向错误进行了详细分析。尽管基于 trio 的使用 HiFi 的方法是当前的黄金标准,但使用 Strand-seq 而不是亲本数据时,染色体级别的相位准确性相当。重要的是,大多数组装缺口聚集在最大和最相似的重复序列附近(包括片段重复[35.4%]、卫星 DNA [22.3%]或富含 GA/AT 丰富 DNA 的区域[27.4%])。因此,至少有 1513 个蛋白编码基因在至少一个单体型中重叠组装缺口,并且有 231 个基因经常从五个或更多单体型中断裂或缺失。此外,我们估计每个单体型有 6-7 Mbp 的 DNA 定向错误,无论是否使用无 trio 或基于 trio 的方法。在这些定向错误中,81%对应于人类物种中真正的大型倒位多态性,其中大多数被大片段重复序列包围。我们还确定了与每个单体基因组 11.9 Mbp 的缺失和 161.4 Mbp 的插入相一致的大规模对齐不连续性。尽管这种变异的 99%对应于卫星 DNA,但我们鉴定出 230 个常染色质 DNA 区域具有频繁的扩展和收缩,其中近一半与 197 个蛋白编码基因重叠。这种可变的和不完全组装的区域是未来算法开发和泛基因组表示的重要目标。