Tuberculosis Genomics Unit, Instituto de Biomedicina de Valencia, Consejo Superior de Investigaciones Científicas, Valencia, Spain.
Centro de Investigação em Saúde de Manhiça, Maputo, Mozambique.
Lancet Glob Health. 2023 Jun;11(6):e933-e941. doi: 10.1016/S2214-109X(23)00169-9.
From the start of the SARS-CoV-2 outbreak, global sequencing efforts have generated an unprecedented amount of genomic data. Nonetheless, unequal sampling between high-income and low-income countries hinders the implementation of genomic surveillance systems at the global and local level. Filling the knowledge gaps of genomic information and understanding pandemic dynamics in low-income countries is essential for public health decision making and to prepare for future pandemics. In this context, we aimed to discover the timing and origin of SARS-CoV-2 variant introductions in Mozambique, taking advantage of pandemic-scale phylogenies.
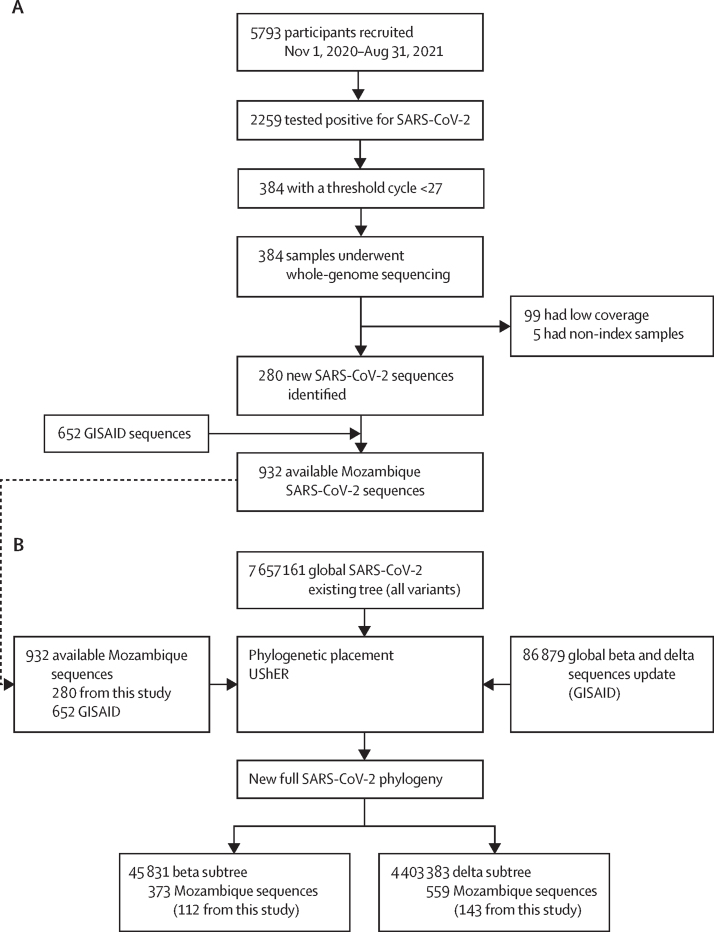
We did a retrospective, observational study in southern Mozambique. Patients from Manhiça presenting with respiratory symptoms were recruited, and those enrolled in clinical trials were excluded. Data were included from three sources: (1) a prospective hospital-based surveillance study (MozCOVID), recruiting patients living in Manhiça, attending the Manhiça district hospital, and fulfilling the criteria of suspected COVID-19 case according to WHO; (2) symptomatic and asymptomatic individuals with SARS-CoV-2 infection recruited by the National Surveillance system; and (3) sequences from SARS-CoV-2-infected Mozambican cases deposited on the Global Initiative on Sharing Avian Influenza Data database. Positive samples amenable for sequencing were analysed. We used Ultrafast Sample placement on Existing tRees to understand the dynamics of beta and delta waves, using available genomic data. This tool can reconstruct a phylogeny with millions of sequences by efficient sample placement in a tree. We reconstructed a phylogeny (~7·6 million sequences) adding new and publicly available beta and delta sequences.
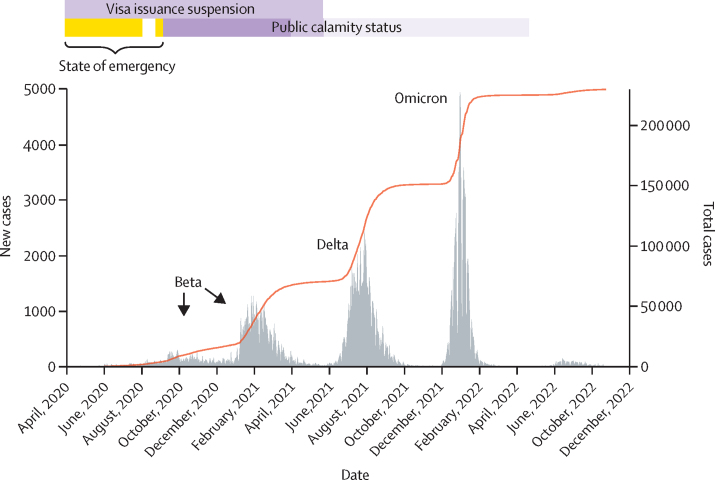
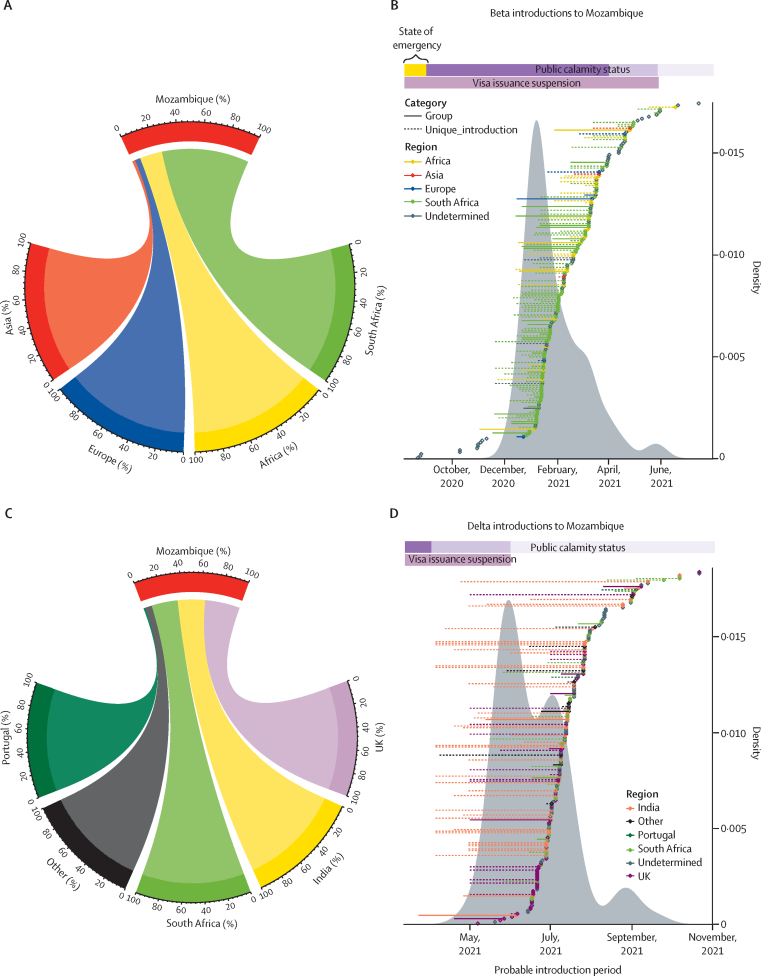
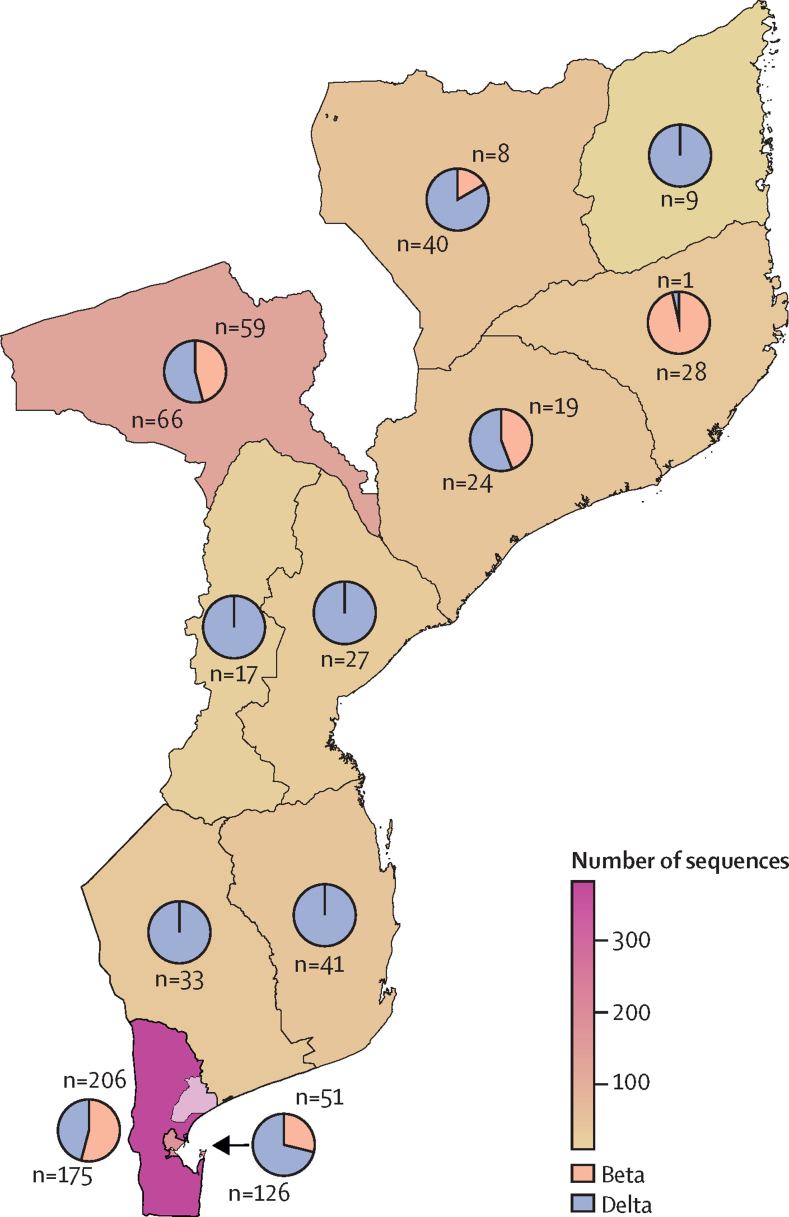
A total of 5793 patients were recruited between Nov 1, 2020, and Aug 31, 2021. During this time, 133 328 COVID-19 cases were reported in Mozambique. 280 good quality new SARS-CoV-2 sequences were obtained after the inclusion criteria were applied and an additional 652 beta (B.1.351) and delta (B.1.617.2) public sequences were included from Mozambique. We evaluated 373 beta and 559 delta sequences. We identified 187 beta introductions (including 295 sequences), divided in 42 transmission groups and 145 unique introductions, mostly from South Africa, between August, 2020 and July, 2021. For delta, we identified 220 introductions (including 494 sequences), with 49 transmission groups and 171 unique introductions, mostly from the UK, India, and South Africa, between April and November, 2021.
The timing and origin of introductions suggests that movement restrictions effectively avoided introductions from non-African countries, but not from surrounding countries. Our results raise questions about the imbalance between the consequences of restrictions and health benefits. This new understanding of pandemic dynamics in Mozambique can be used to inform public health interventions to control the spread of new variants.
European and Developing Countries Clinical Trials, European Research Council, Bill & Melinda Gates Foundation, and Agència de Gestió d'Ajuts Universitaris i de Recerca.
自 SARS-CoV-2 疫情爆发以来,全球测序工作已经产生了前所未有的大量基因组数据。然而,高收入和低收入国家之间的采样不均衡阻碍了全球和地方层面基因组监测系统的实施。填补基因组信息的知识空白,了解低收入国家的大流行动态,对于公共卫生决策和为未来的大流行做好准备至关重要。在这种情况下,我们旨在利用大流行规模的系统发育学来发现 SARS-CoV-2 变异在莫桑比克的引入时间和起源。
我们在莫桑比克南部进行了一项回顾性观察性研究。招募了来自马希齐出现呼吸道症状的患者,并排除了参加临床试验的患者。数据来自三个来源:(1)在马希齐进行的一项基于医院的前瞻性监测研究(MozCOVID),招募居住在马希齐、在马希齐区医院就诊并根据世界卫生组织标准符合疑似 COVID-19 病例标准的患者;(2)国家监测系统招募的有症状和无症状 SARS-CoV-2 感染个体;以及(3)在全球共享禽流感数据倡议数据库中登记的感染 SARS-CoV-2 的莫桑比克病例的序列。分析了适合测序的阳性样本。我们使用现有树上的超快样本放置来理解贝塔和德尔塔波的动态,利用可用的基因组数据。该工具可以通过在树中有效放置样本,重建具有数百万个序列的系统发育。我们重建了一个系统发育(约 760 万个序列),增加了新的和公开的贝塔和德尔塔序列。
2020 年 11 月 1 日至 2021 年 8 月 31 日期间,共招募了 5793 名患者。在此期间,莫桑比克报告了 133328 例 COVID-19 病例。在应用纳入标准后,获得了 280 个高质量的新 SARS-CoV-2 序列,并从莫桑比克获得了另外 652 个贝塔(B.1.351)和德尔塔(B.1.617.2)公共序列。我们评估了 373 个贝塔和 559 个德尔塔序列。我们发现了 187 次贝塔引入(包括 295 个序列),分为 42 个传播组和 145 个独特引入,主要来自南非,时间在 2020 年 8 月至 2021 年 7 月之间。对于德尔塔,我们发现了 220 次引入(包括 494 个序列),分为 49 个传播组和 171 个独特引入,主要来自英国、印度和南非,时间在 2021 年 4 月至 11 月之间。
引入的时间和起源表明,限制流动有效地避免了来自非非洲国家的引入,但不能避免来自周边国家的引入。我们的结果提出了关于限制措施的后果和健康益处之间不平衡的问题。对莫桑比克大流行动态的这种新理解可以用于为控制新变体的传播提供公共卫生干预措施。
欧洲和发展中国家临床试验、欧洲研究理事会、比尔及梅琳达盖茨基金会和 Agència de Gestió d'Ajuts Universitaris i de Recerca。