Department of Chemical Engineering, Military Institute of Engineering, Rio de Janeiro, RJ, Brazil.
Laboratory of Molecular Modeling Applied to Chemical and Biological Defense (LMCBD), Military Institute of Engineering, Rio de Janeiro, RJ, Brazil.
J Mol Model. 2023 May 22;29(6):183. doi: 10.1007/s00894-023-05586-5.
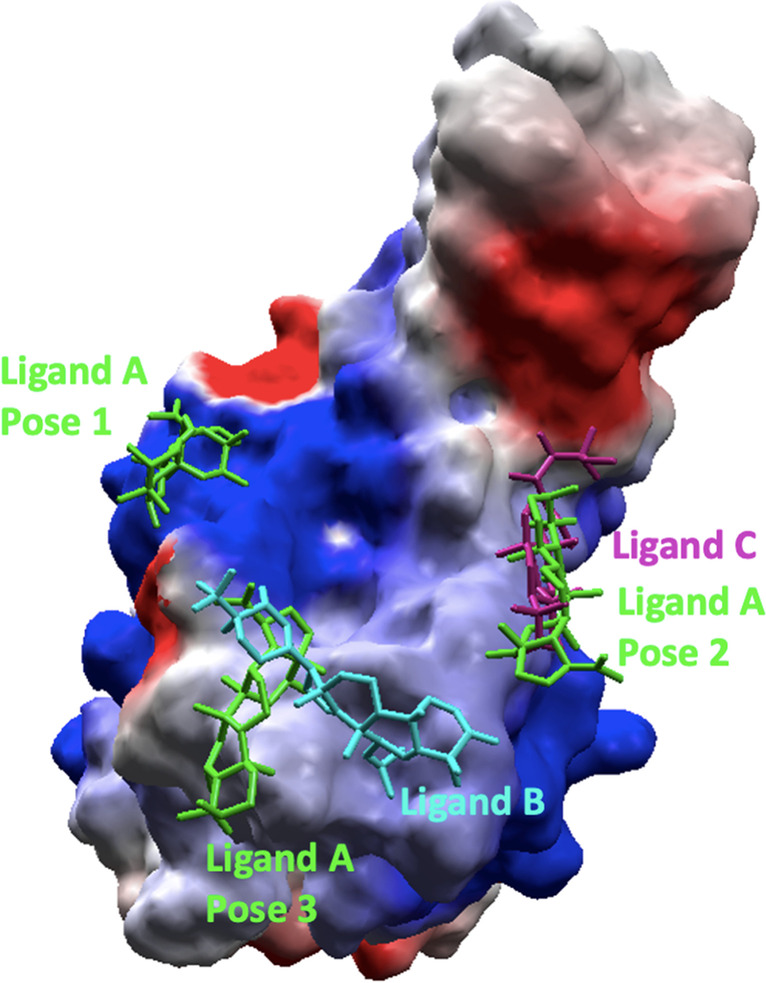
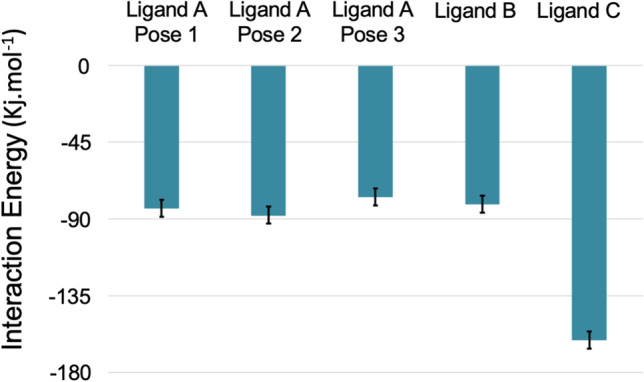
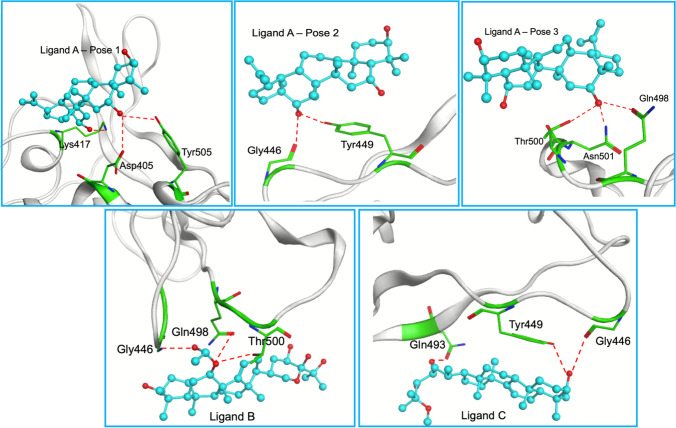
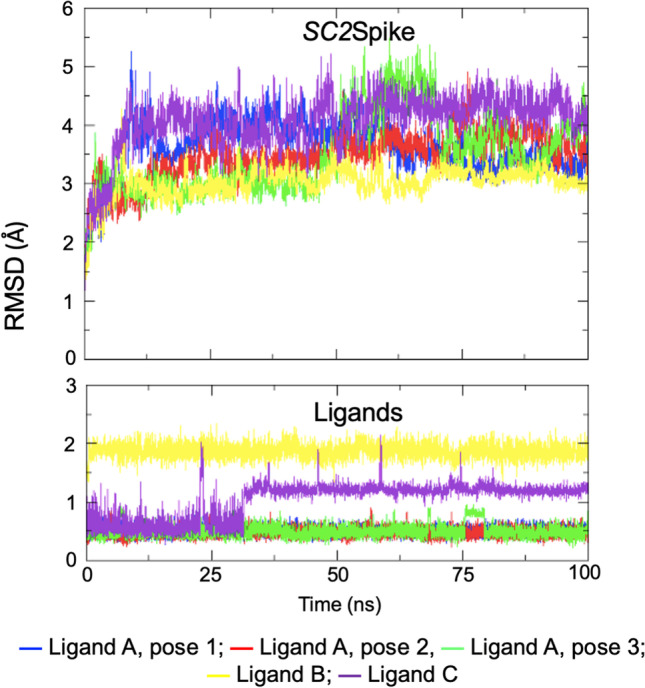
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the COVID-19 infection and responsible for millions of victims worldwide, remains a significant threat to public health. Even after the development of vaccines, research interest in the emergence of new variants is still prominent. Currently, the focus is on the search for effective and safe drugs, given the limitations and side effects observed for the synthetic drugs administered so far. In this sense, bioactive natural products that are widely used in the pharmaceutical industry due to their effectiveness and low toxicity have emerged as potential options in the search for safe drugs against COVID-19. Following this line, we screened 10 bioactive compounds derived from cholesterol for molecules capable of interacting with the receptor-binding domain (RBD) of the spike protein from SARS-CoV-2 (SC2Spike), responsible for the virus's invasion of human cells. Rounds of docking followed by molecular dynamics simulations and binding energy calculations enabled the selection of three compounds worth being experimentally evaluated against SARS-CoV-2.
The 3D structures of the cholesterol derivatives were prepared and optimized using the Spartan 08 software with the semi-empirical method PM3. They were then exported to the Molegro Virtual Docking (MVD®) software, where they were docked onto the RBD of a 3D structure of the SC2Spike protein that was imported from the Protein Data Bank (PDB). The best poses obtained from MVD® were subjected to rounds of molecular dynamics simulations using the GROMACS software, with the OPLS/AA force field. Frames from the MD simulation trajectories were used to calculate the ligand's free binding energies using the molecular mechanics - Poisson-Boltzmann surface area (MM-PBSA) method. All results were analyzed using the xmgrace and Visual Molecular Dynamics (VMD) software.
严重急性呼吸系统综合征冠状病毒 2(SARS-CoV-2)是 COVID-19 感染的病原体,也是导致全球数百万人患病的罪魁祸首,它仍然是对公共健康的重大威胁。即使开发了疫苗,对新变体出现的研究兴趣仍然很突出。目前,鉴于迄今为止给予的合成药物的局限性和副作用,重点是寻找有效和安全的药物。从这个意义上说,由于其有效性和低毒性而在制药工业中广泛使用的生物活性天然产物已成为寻找安全的 COVID-19 药物的潜在选择。沿着这条线,我们筛选了 10 种源自胆固醇的生物活性化合物,以寻找能够与 SARS-CoV-2(SC2Spike)刺突蛋白的受体结合域(RBD)相互作用的分子,该蛋白负责病毒入侵人体细胞。经过几轮对接,随后进行分子动力学模拟和结合能计算,选择了三种值得针对 SARS-CoV-2 进行实验评估的化合物。
使用 Spartan 08 软件和半经验 PM3 方法对胆固醇衍生物的 3D 结构进行准备和优化。然后将它们导出到 Molegro Virtual Docking(MVD®)软件中,在该软件中将它们对接在从蛋白质数据银行(PDB)导入的 SC2Spike 蛋白的 3D 结构的 RBD 上。从 MVD®获得的最佳构象经过使用 GROMACS 软件和 OPLS/AA 力场的几轮分子动力学模拟。从 MD 模拟轨迹的帧中使用分子力学 - 泊松-玻尔兹曼表面面积(MM-PBSA)方法计算配体的自由结合能。使用 xmgrace 和 Visual Molecular Dynamics(VMD)软件分析所有结果。