Alghamdi Mansour A, Al-Eitan Laith N, Tarkhan Amneh H
Department of Anatomy, College of Medicine, King Khalid University, Abha, 61421, Saudi Arabia.
Genomics and Personalized Medicine Unit, College of Medicine, King Khalid University, Abha, 61421, Saudi Arabia.
Heliyon. 2023 May 7;9(5):e16101. doi: 10.1016/j.heliyon.2023.e16101. eCollection 2023 May.
Human papillomaviruses have been shown to dysregulate the gene expression and DNA methylation profiles of their host cells over the course of infection. However, there is a lack of information on the impact of low-risk HPV infection and wart formation on host cell's expression and methylation patterns. Therefore, the objective of this study is to analyse the genome and methylome of common warts using an integrative approach.
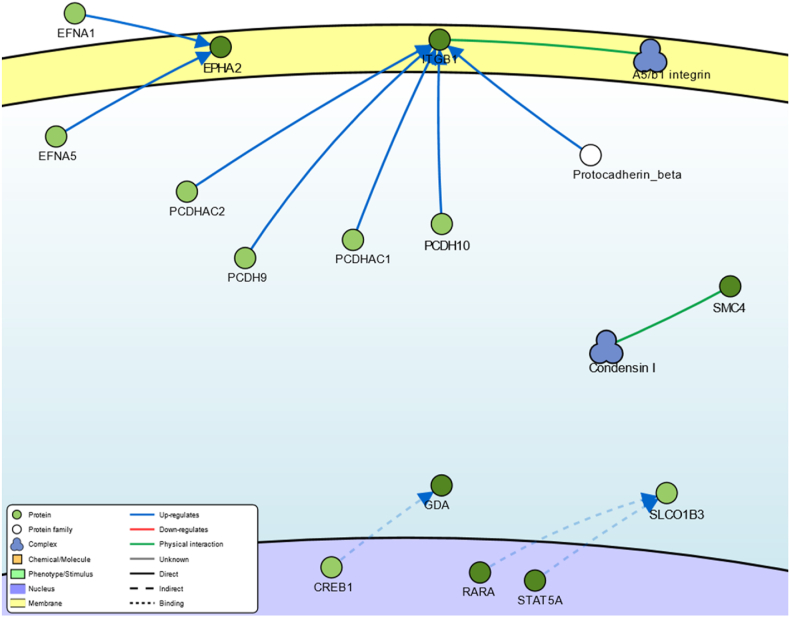
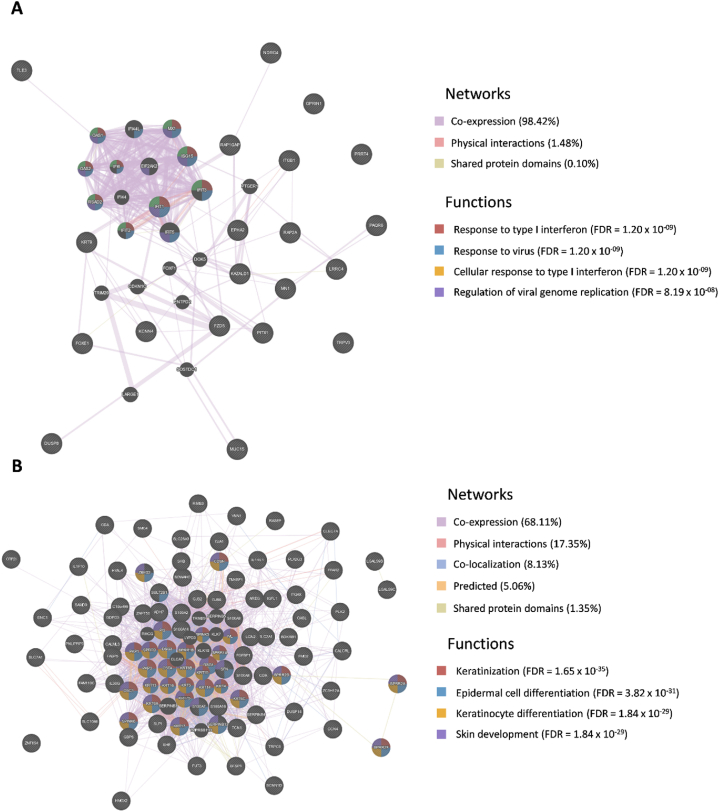
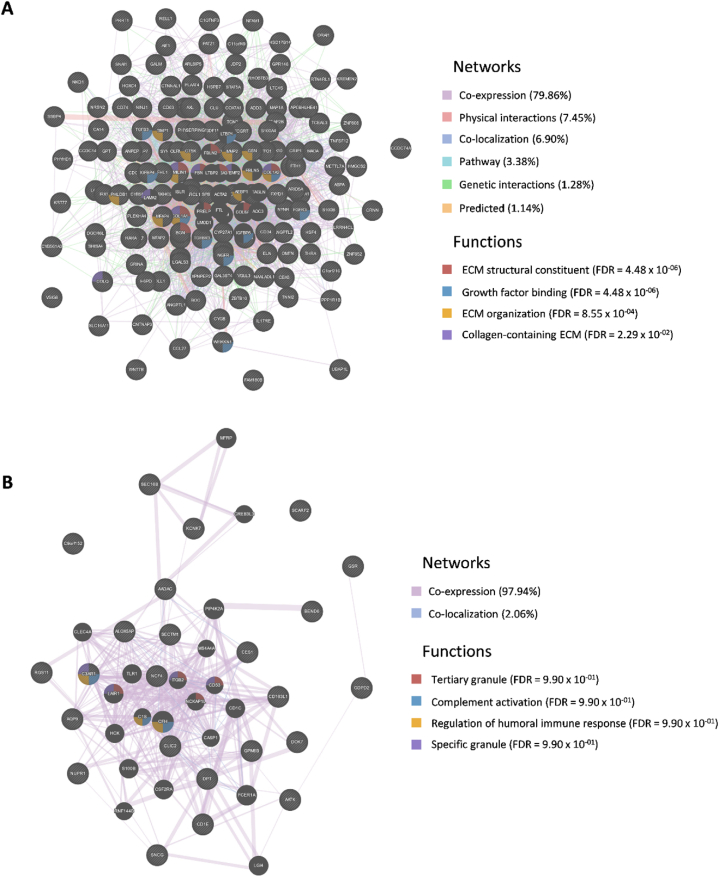
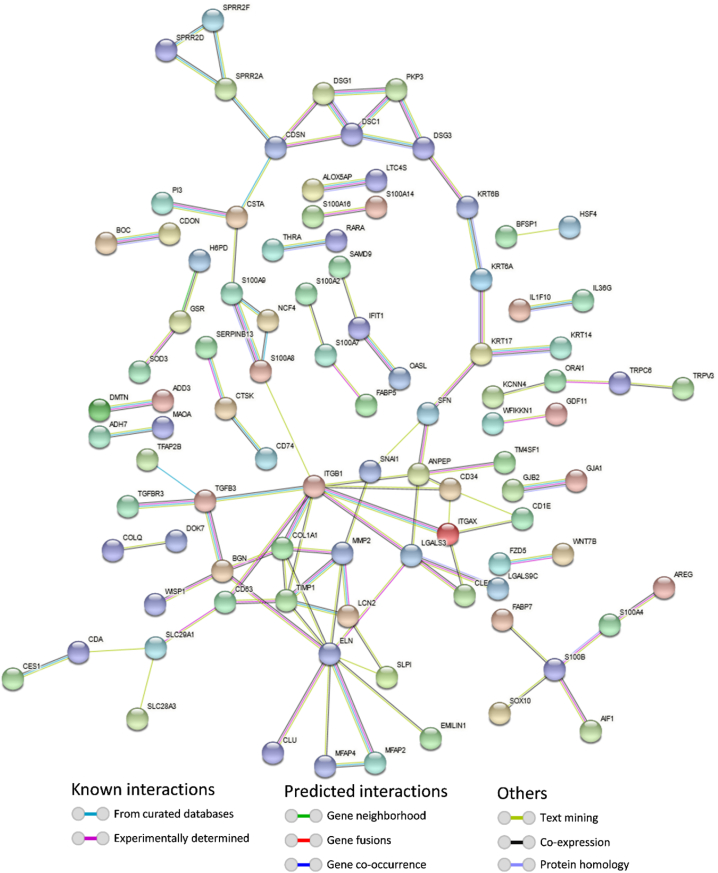
In the present study, gene expression (GSE136347) and methylation (GSE213888) datasets of common warts were obtained from the GEO database. Identification of the differentially expressed and differentially methylated genes was carried out using the RnBeads R package and the edgeR Bioconductor package. Next, functional annotation of the identified genes was obtained using the Database for Annotation, Visualization, and Integrated Discovery (DAVID). Network construction and analyses of the gene-gene, protein-protein, and signaling interactions of the differentially expressed and differentially methylated genes was performed using the GeneMANIA web interface, the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database, and the Signaling Network Open Resource 2.0 (SIGNOR 2.0), respectively. Lastly, significant hub genes were identified using the Cytoscape application CytoHubba.
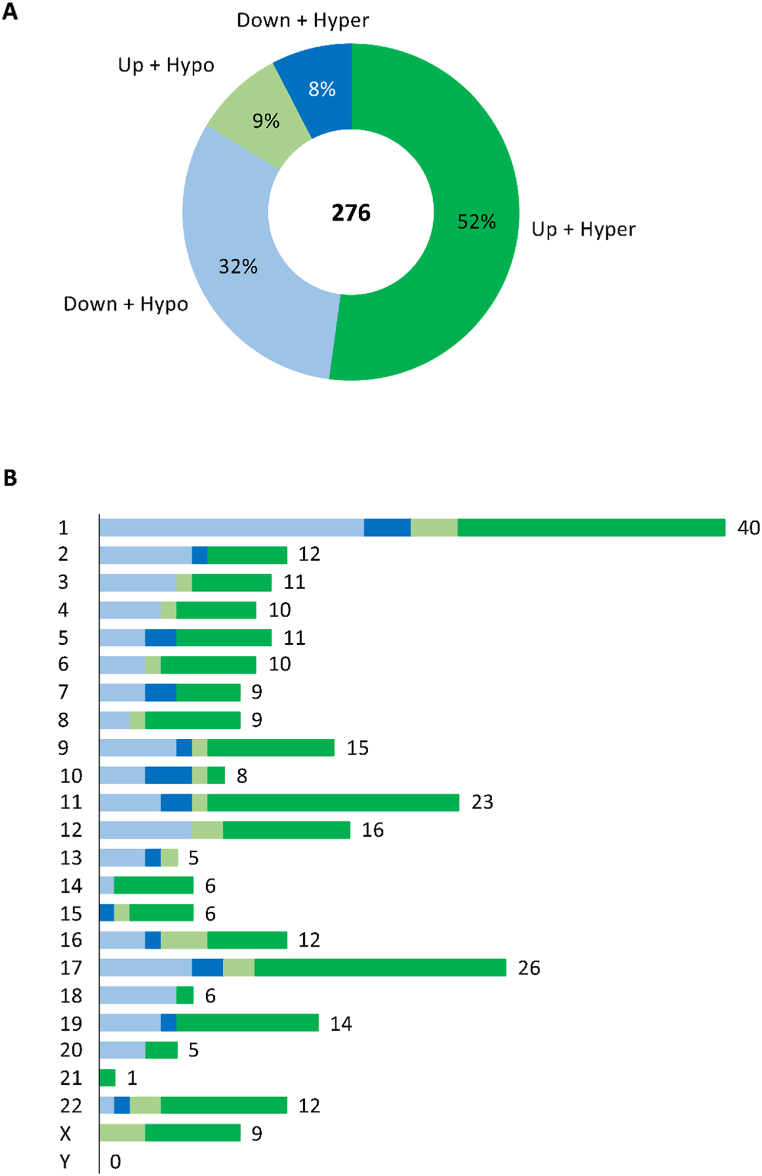
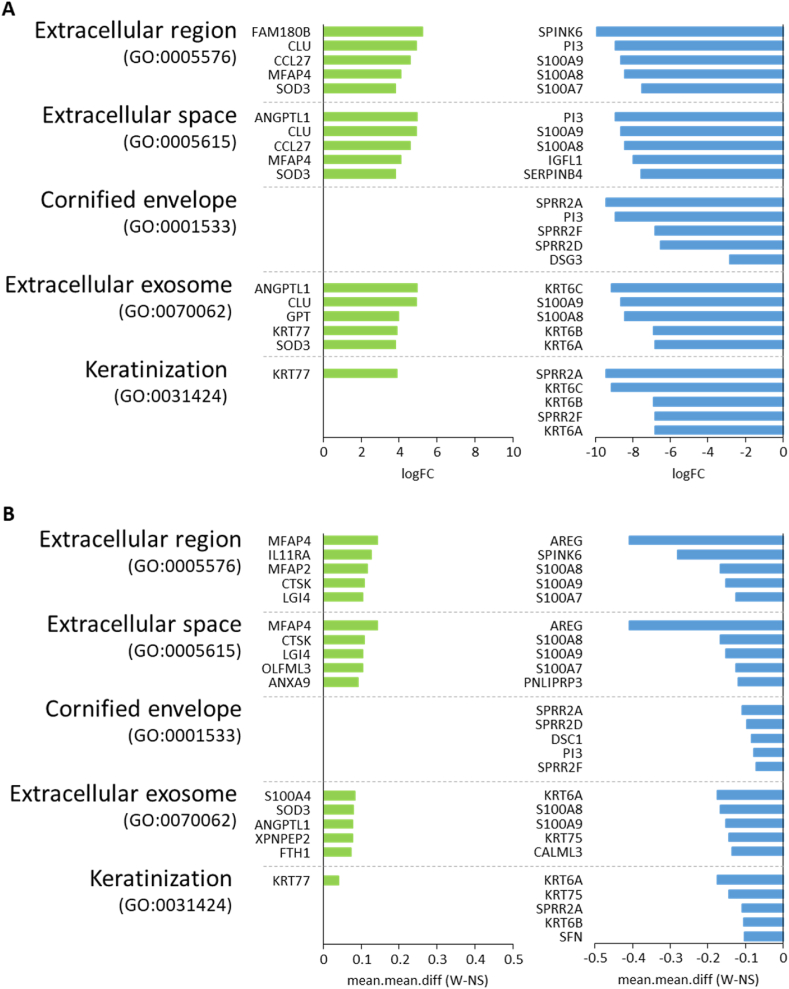
A total of 276 genes were identified as differentially expressed and differentially methylated in common warts, with 52% being upregulated and hypermethylated. Functional enrichment analysis identified extracellular components as the most enriched annotations, while network analyses identified and as significant hub genes.
To the best knowledge of the authors, this is the first integrative study to be carried out on non-genital warts induced by low-risk HPV types. Future studies are required to re-validate the findings in larger populations using alternative approaches.
人乳头瘤病毒已被证明在感染过程中会失调其宿主细胞的基因表达和DNA甲基化谱。然而,关于低风险HPV感染和疣形成对宿主细胞表达和甲基化模式的影响,目前缺乏相关信息。因此,本研究的目的是使用综合方法分析寻常疣的基因组和甲基化组。
在本研究中,从GEO数据库获得寻常疣的基因表达(GSE136347)和甲基化(GSE213888)数据集。使用RnBeads R包和edgeR Bioconductor包进行差异表达基因和差异甲基化基因的鉴定。接下来,使用注释、可视化和综合发现数据库(DAVID)对鉴定出的基因进行功能注释。分别使用GeneMANIA网络界面、相互作用基因/蛋白质检索工具(STRING)数据库和信号网络开放资源2.0(SIGNOR 2.0)对差异表达基因和差异甲基化基因的基因-基因、蛋白质-蛋白质和信号相互作用进行网络构建和分析。最后,使用Cytoscape应用程序CytoHubba鉴定重要的枢纽基因。
共鉴定出276个在寻常疣中差异表达和差异甲基化的基因,其中52%的基因上调且高甲基化。功能富集分析确定细胞外成分是最丰富的注释,而网络分析确定 和 为重要的枢纽基因。
据作者所知,这是首次对低风险HPV类型引起的非生殖器疣进行的综合研究。未来需要使用替代方法在更大的人群中重新验证这些发现。