Science for Life Laboratory & Swedish e-Science Research Center, Department of Applied Physics, KTH Royal Institute of Technology, Solna, Sweden.
Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, Solna, Sweden.
Biophys J. 2023 Jul 11;122(13):2773-2781. doi: 10.1016/j.bpj.2023.05.033. Epub 2023 Jun 5.
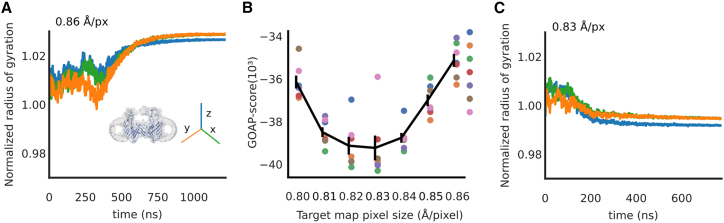
The resolution revolution has increasingly enabled single-particle cryogenic electron microscopy (cryo-EM) reconstructions of previously inaccessible systems, including membrane proteins-a category that constitutes a disproportionate share of drug targets. We present a protocol for using density-guided molecular dynamics simulations to automatically refine atomistic models into membrane protein cryo-EM maps. Using adaptive force density-guided simulations as implemented in the GROMACS molecular dynamics package, we show how automated model refinement of a membrane protein is achieved without the need to manually tune the fitting force ad hoc. We also present selection criteria to choose the best-fit model that balances stereochemistry and goodness of fit. The proposed protocol was used to refine models into a new cryo-EM density of the membrane protein maltoporin, either in a lipid bilayer or detergent micelle, and we found that results do not substantially differ from fitting in solution. Fitted structures satisfied classical model-quality metrics and improved the quality and the model-to-map correlation of the x-ray starting structure. Additionally, the density-guided fitting in combination with generalized orientation-dependent all-atom potential was used to correct the pixel-size estimation of the experimental cryo-EM density map. This work demonstrates the applicability of a straightforward automated approach to fitting membrane protein cryo-EM densities. Such computational approaches promise to facilitate rapid refinement of proteins under different conditions or with various ligands present, including targets in the highly relevant superfamily of membrane proteins.
分辨率革命已经越来越能够对以前无法获得的系统进行单颗粒低温电子显微镜(cryo-EM)重建,包括膜蛋白——这一类别构成了不成比例的药物靶点份额。我们提出了一种使用密度引导的分子动力学模拟自动将原子模型细化到膜蛋白 cryo-EM 图谱中的方案。我们使用在 GROMACS 分子动力学包中实现的自适应力密度引导模拟,展示了如何在无需手动调整拟合力的情况下自动细化膜蛋白模型。我们还提出了选择最佳拟合模型的标准,该模型平衡立体化学和拟合度。所提出的方案用于将模型细化到膜蛋白麦芽糖结合蛋白的新 cryo-EM 密度中,无论是在脂质双层还是去污剂胶束中,我们发现结果与在溶液中的拟合没有实质性差异。拟合结构满足经典的模型质量指标,并提高了 X 射线起始结构的质量和模型与图谱的相关性。此外,密度引导拟合与广义方向相关的全原子势能结合使用,可纠正实验 cryo-EM 密度图谱的像素尺寸估计。这项工作证明了一种简单的自动方法适用于拟合膜蛋白 cryo-EM 密度。这种计算方法有望促进不同条件下或存在各种配体的蛋白质的快速细化,包括在高度相关的膜蛋白超家族中的靶标。