Department of Human Stress Response Science, Institute of Biomedical Science, Kansai Medical University, 2-5-1 Shin-machi, Hirakata, Osaka, 573-1010, Japan.
Department of Anesthesiology and Intensive Care, Osaka Red Cross Hospital, Osaka, Japan.
Sci Rep. 2023 Jun 16;13(1):9785. doi: 10.1038/s41598-023-37016-0.
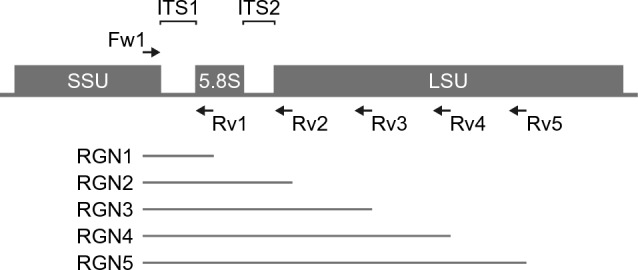
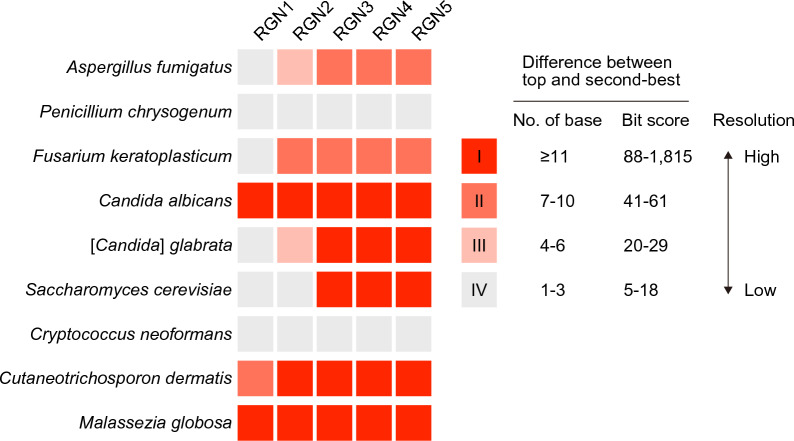
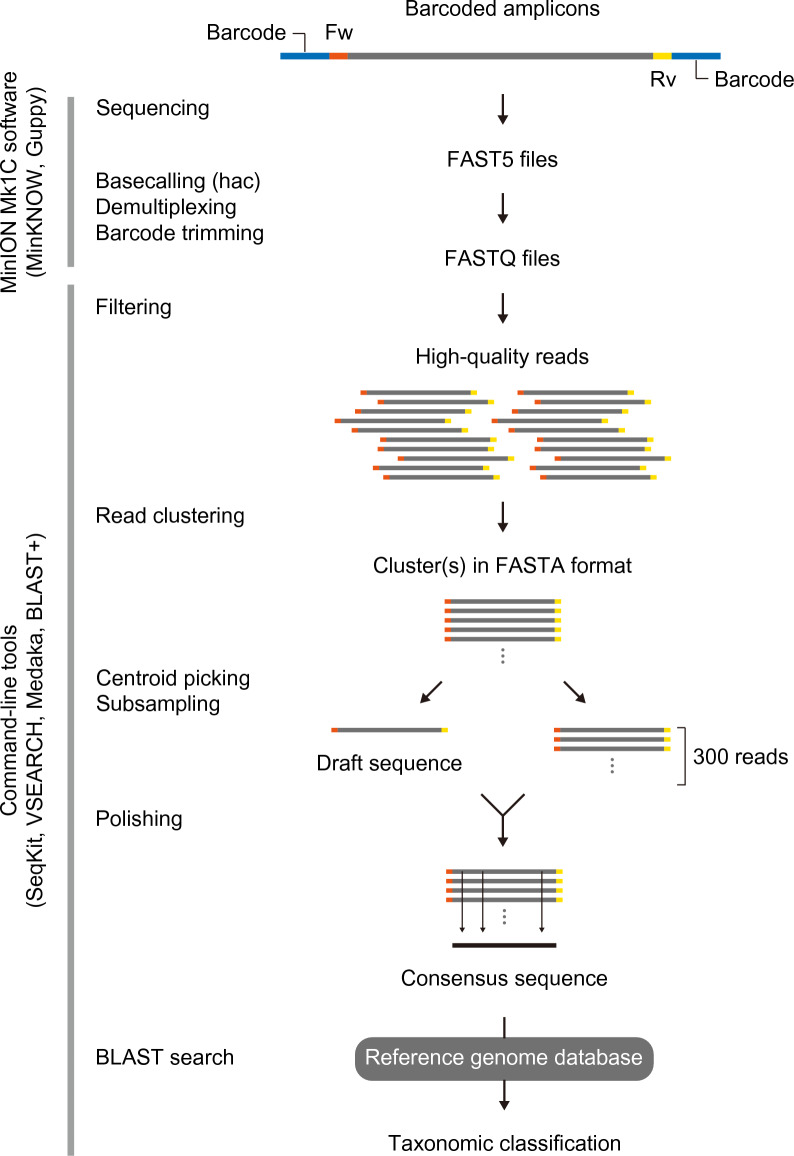
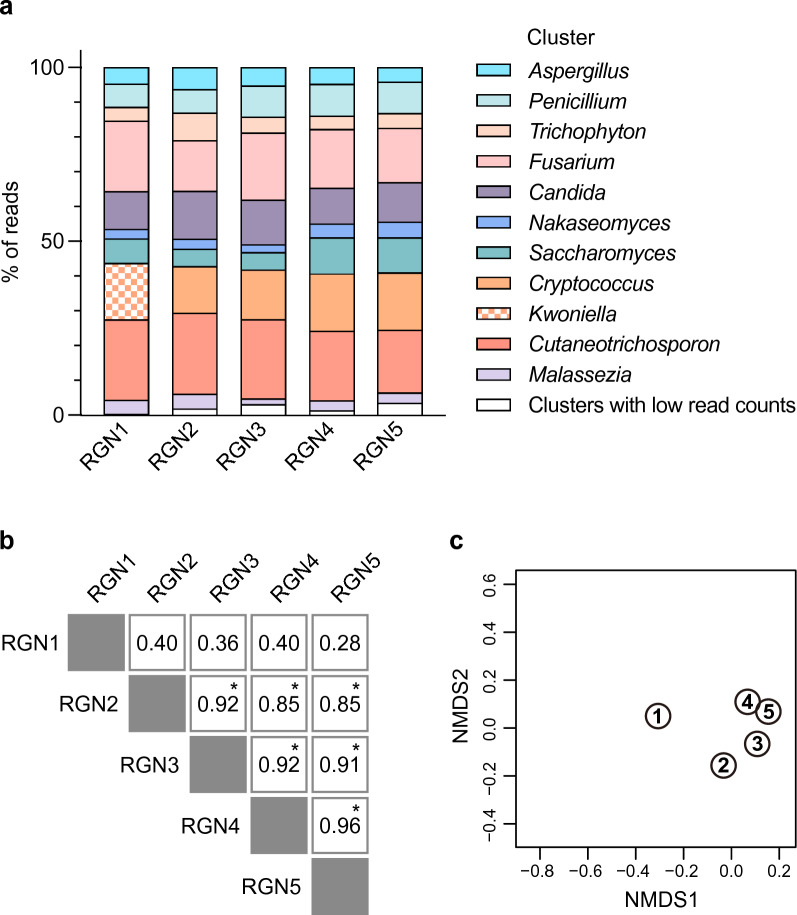
The study of microbiota has been revolutionized by the development of DNA metabarcoding. This sequence-based approach enables the direct detection of microorganisms without the need for culture and isolation, which significantly reduces analysis time and offers more comprehensive taxonomic profiles across broad phylogenetic lineages. While there has been an accumulating number of researches on bacteria, molecular phylogenetic analysis of fungi still remains challenging due to the lack of standardized tools and the incompleteness of reference databases limiting the accurate and precise identification of fungal taxa. Here, we present a DNA metabarcoding workflow for characterizing fungal microbiota with high taxonomic resolution. This method involves amplifying longer stretches of ribosomal RNA operons and sequencing them using nanopore long-read sequencing technology. The resulting reads were error-polished to generate consensus sequences with 99.5-100% accuracy, which were then aligned against reference genome assemblies. The efficacy of this method was explored using a polymicrobial mock community and patient-derived specimens, demonstrating the marked potential of long-read sequencing combined with consensus calling for accurate taxonomic classification. Our approach offers a powerful tool for the rapid identification of pathogenic fungi and has the promise to significantly improve our understanding of the role of fungi in health and disease.
微生物组学的研究已经被 DNA 代谢组学的发展所改变。这种基于序列的方法可以直接检测微生物,而无需培养和分离,这大大减少了分析时间,并在广泛的系统发育谱系中提供了更全面的分类群概况。虽然关于细菌的研究越来越多,但由于缺乏标准化的工具和参考数据库的不完整性,真菌的分子系统发育分析仍然具有挑战性,这限制了真菌分类群的准确和精确识别。在这里,我们提出了一种具有高分类分辨率的真菌微生物组 DNA 代谢组学工作流程。该方法涉及扩增更长的核糖体 RNA 操纵子片段,并使用纳米孔长读测序技术对其进行测序。将得到的reads 进行错误修正,以生成具有 99.5-100%准确率的共识序列,然后将其与参考基因组组装进行比对。使用多微生物模拟群落和患者来源的标本探索了这种方法的效果,证明了长读测序与共识调用相结合用于准确分类的显著潜力。我们的方法为快速鉴定致病性真菌提供了一种强大的工具,并有望显著提高我们对真菌在健康和疾病中的作用的理解。