Key Laboratory of Arrhythmias of the Ministry of Education of China, Research Center for Translational Medicine, Shanghai Heart Failure Research Center, Department of Cardiovascular Surgery, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, 200120, China.
Key Laboratory of Arrhythmias of the Ministry of Education of China, Research Center for Translational Medicine, Shanghai Heart Failure Research Center, Department of Cardiovascular Surgery, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, 200120, China; Shenzhen Ruipuxun Academy for Stem Cell and Regenerative Medicine, Shenzhen, China.
Redox Biol. 2023 Aug;64:102775. doi: 10.1016/j.redox.2023.102775. Epub 2023 Jun 12.
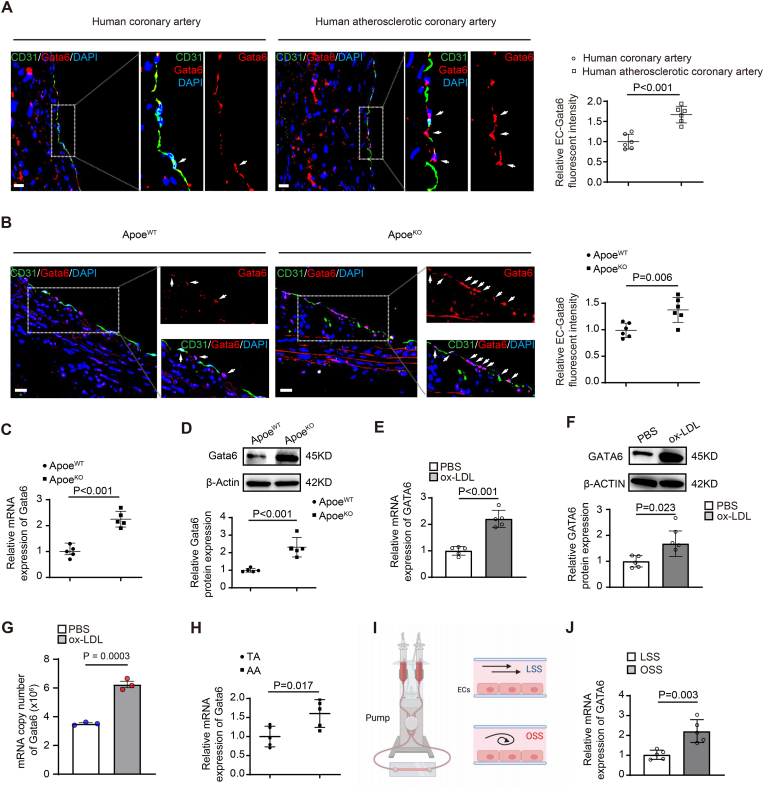
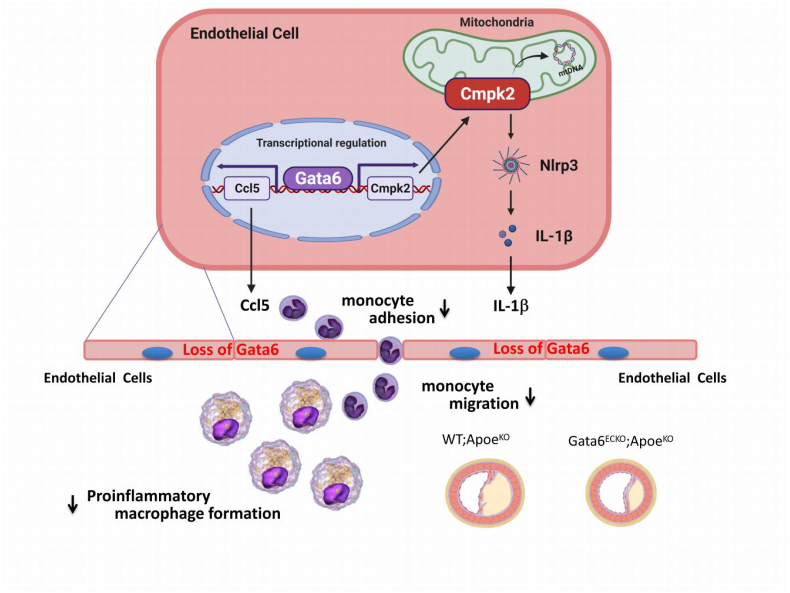
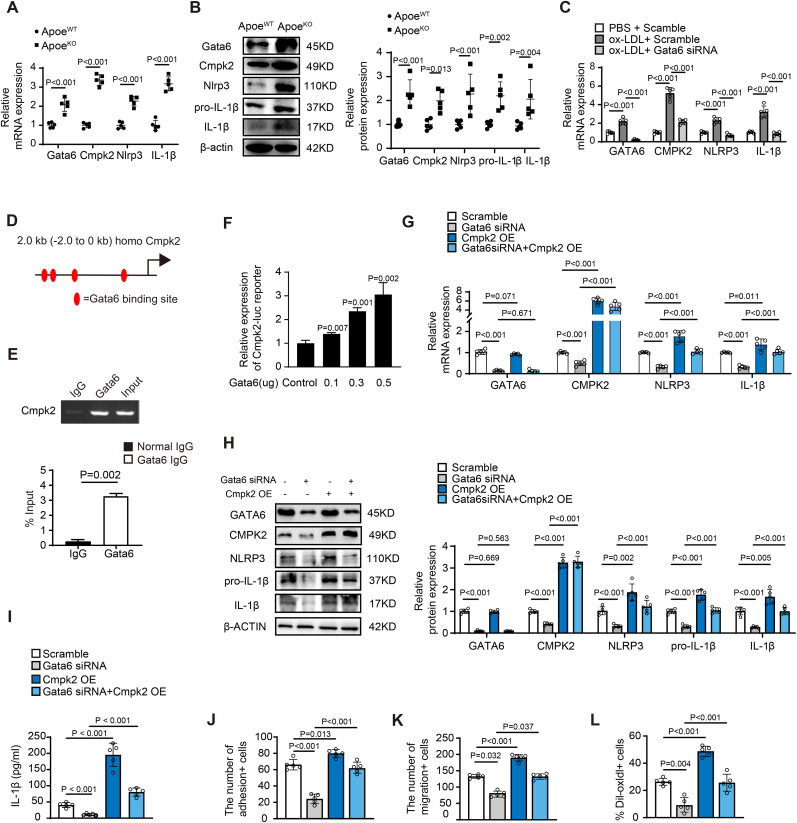
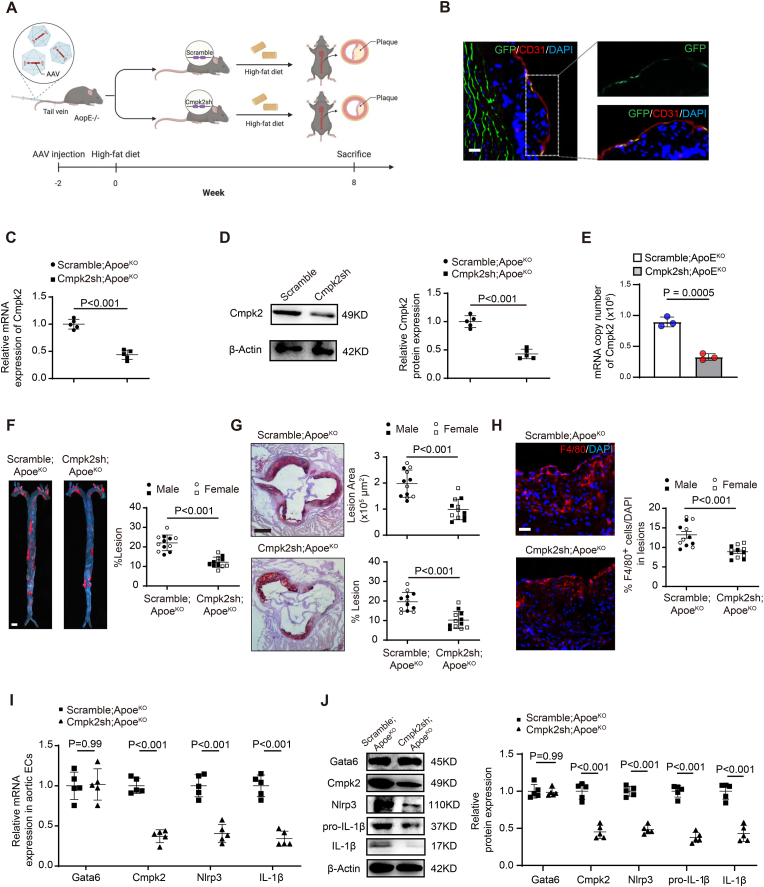
Endothelial dysfunction results in chronic vascular inflammation, which is critical for the development of atherosclerotic diseases. Transcription factor Gata6 has been reported to regulate vascular endothelial cell activation and inflammation in vitro. Here, we aimed to explore the roles and mechanisms of endothelial Gata6 in atherogenesis. Endothelial cell (EC) specific Gata6 deletion was generated in the Apoe hyperlipidemic atherosclerosis mouse model. Atherosclerotic lesion formation, endothelial inflammatory signaling, and endothelial-macrophage interaction were examined in vivo and in vitro by using cellular and molecular biological approaches. EC-GATA6 deletion mice exhibited a significant decrease in monocyte infiltration and atherosclerotic lesion compared to littermate control mice. Cytosine monophosphate kinase 2 (Cmpk2) was identified as a direct target gene of GATA6 and EC-GATA6 deletion decreased monocyte adherence, migration and pro-inflammatory macrophage foam cell formation through regulation of the CMPK2-Nlrp3 pathway. Endothelial target delivery of Cmpk2-shRNA by intercellular adhesion molecule 2 (Icam-2) promoter-driven AAV9 carrying the shRNA reversed the Gata6 upregulation mediated elevated Cmpk2 expression and further Nlrp3 activation and thus attenuated atherosclerosis. In addition, C-C motif chemokine ligand 5 (Ccl5) was also identified as a direct target gene of Gata6 to regulate monocyte adherence and migration influencing atherogenesis. This study provides direct in vivo evidence of EC-GATA6 involvement in the regulation of Cmpk2-Nlrp3, as well as Ccl5, on monocyte adherence and migration in atherosclerosis development and advances our understanding of the in vivo mechanisms of atherosclerotic lesion development, and meanwhile provides opportunities for future therapeutic interventions.
内皮功能障碍导致慢性血管炎症,这对于动脉粥样硬化疾病的发展至关重要。转录因子 Gata6 已被报道可调节体外血管内皮细胞的激活和炎症。在这里,我们旨在探讨内皮 Gata6 在动脉粥样硬化形成中的作用和机制。在 Apoe 高脂血症动脉粥样硬化小鼠模型中生成内皮细胞(EC)特异性 Gata6 缺失。通过细胞和分子生物学方法在体内和体外研究了内皮 Gata6 缺失对动脉粥样硬化病变形成、内皮炎症信号和内皮-巨噬细胞相互作用的影响。与同窝对照小鼠相比,EC-GATA6 缺失小鼠单核细胞浸润和动脉粥样硬化病变明显减少。环磷酸鸟苷激酶 2(Cmpk2)被鉴定为 GATA6 的直接靶基因,EC-GATA6 缺失通过调节 CMPK2-Nlrp3 通路降低单核细胞黏附、迁移和促炎巨噬细胞泡沫细胞形成。通过细胞间黏附分子 2(Icam-2)启动子驱动的携带 shRNA 的 AAV9 进行内皮细胞靶向递送 Cmpk2-shRNA,逆转了 Gata6 上调介导的 Cmpk2 表达升高以及进一步的 Nlrp3 激活,从而减轻了动脉粥样硬化。此外,C-C 基序趋化因子配体 5(Ccl5)也被鉴定为 Gata6 的直接靶基因,以调节单核细胞黏附和迁移,影响动脉粥样硬化的发生。本研究提供了内皮 GATA6 参与调节 Cmpk2-Nlrp3 以及 Ccl5 影响单核细胞黏附和迁移的直接体内证据,阐明了动脉粥样硬化病变发展的体内机制,同时为未来的治疗干预提供了机会。