Department of Otorhinolaryngology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China.
Division of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China.
Int J Mol Sci. 2023 Jun 19;24(12):10349. doi: 10.3390/ijms241210349.
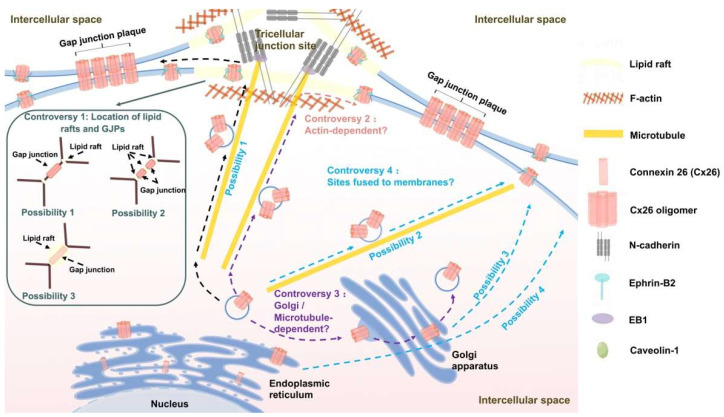
The connexin gene family is the most prevalent gene that contributes to hearing loss. Connexins 26 and 30, encoded by and , respectively, are the most abundantly expressed connexins in the inner ear. Connexin 43, which is encoded by , appears to be widely expressed in various organs, including the heart, skin, the brain, and the inner ear. The mutations that arise in , , and can all result in comprehensive or non-comprehensive genetic deafness in newborns. As it is predicted that connexins include at least 20 isoforms in humans, the biosynthesis, structural composition, and degradation of connexins must be precisely regulated so that the gap junctions can properly operate. Certain mutations result in connexins possessing a faulty subcellular localization, failing to transport to the cell membrane and preventing gap junction formation, ultimately leading to connexin dysfunction and hearing loss. In this review, we provide a discussion of the transport models for connexin 43, connexins 30 and 26, mutations affecting trafficking pathways of these connexins, the existing controversies in the trafficking pathways of connexins, and the molecules involved in connexin trafficking and their functions. This review can contribute to a new way of understanding the etiological principles of connexin mutations and finding therapeutic strategies for hereditary deafness.
缝隙连接蛋白基因家族是导致听力损失最常见的基因。缝隙连接蛋白 26 和 30 分别由 和 编码,是内耳中表达最丰富的缝隙连接蛋白。由 编码的缝隙连接蛋白 43 似乎在多种器官中广泛表达,包括心脏、皮肤、大脑和内耳。在 、 、 和 中发生的突变都可能导致新生儿出现全面或非全面遗传性耳聋。由于预测人类中的缝隙连接蛋白至少包括 20 种同工型,因此必须精确调节缝隙连接蛋白的生物合成、结构组成和降解,以使间隙连接正常工作。某些突变导致缝隙连接蛋白具有错误的亚细胞定位,无法运输到细胞膜并阻止间隙连接形成,最终导致缝隙连接蛋白功能障碍和听力损失。在这篇综述中,我们讨论了缝隙连接蛋白 43、30 和 26 的运输模型、影响这些缝隙连接蛋白运输途径的突变、缝隙连接蛋白运输途径中的现存争议以及参与缝隙连接蛋白运输的分子及其功能。这篇综述可以为理解缝隙连接蛋白突变的病因学原理和寻找遗传性耳聋的治疗策略提供一种新的思路。