National Center for Microscopy and Imaging Research, Center for Research in Biological Systems, University of California San Diego, La Jolla, California, United States of America.
PLoS One. 2013 Aug 15;8(8):e70916. doi: 10.1371/journal.pone.0070916. eCollection 2013.
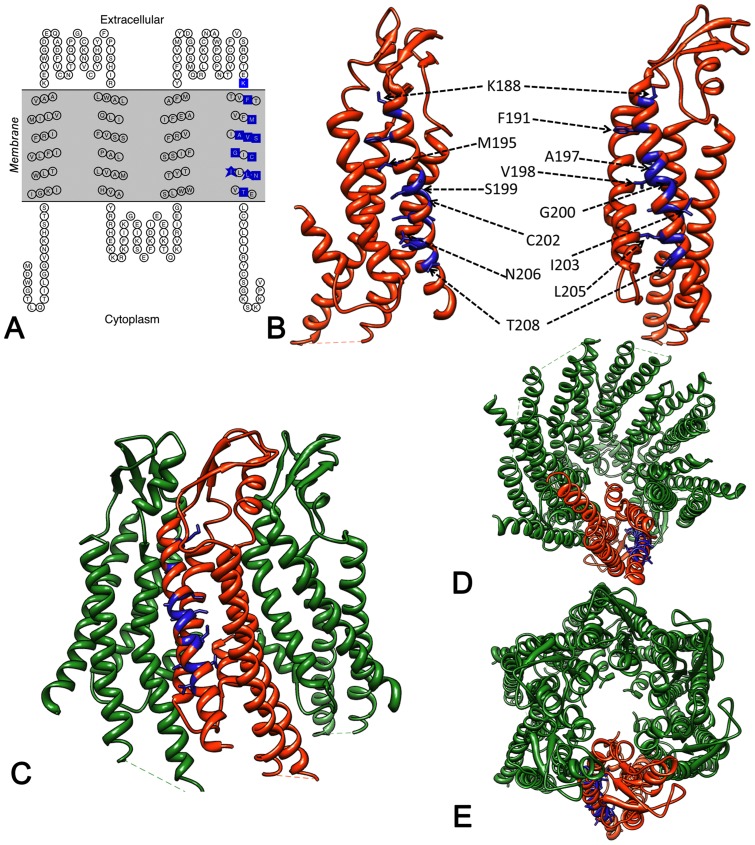
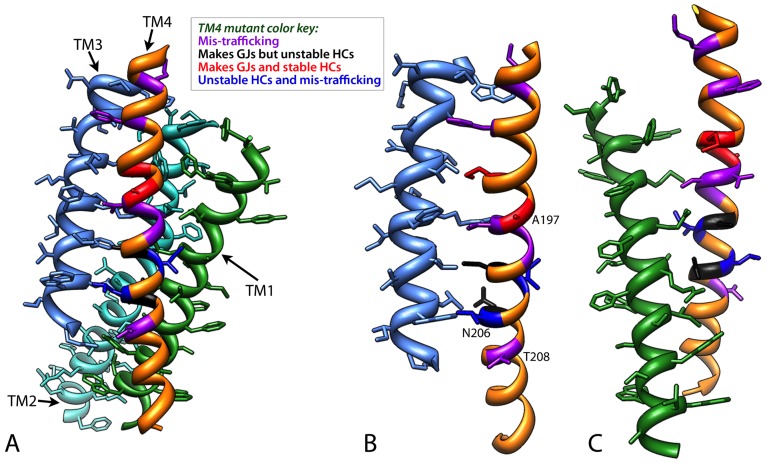
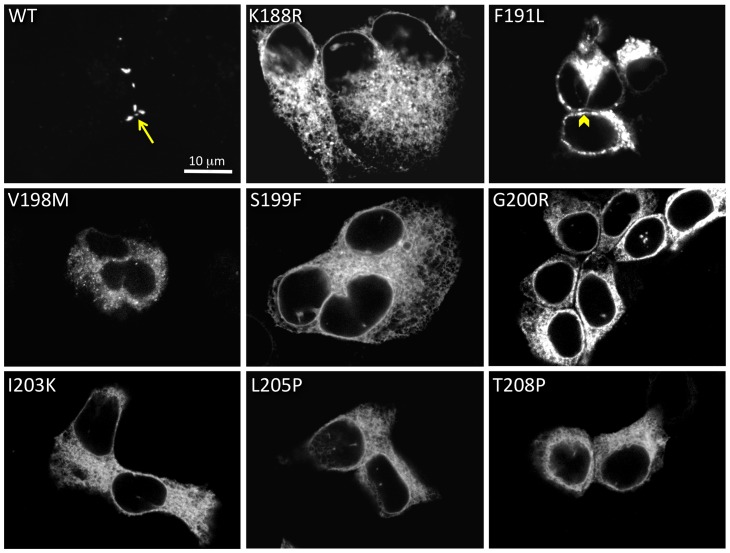
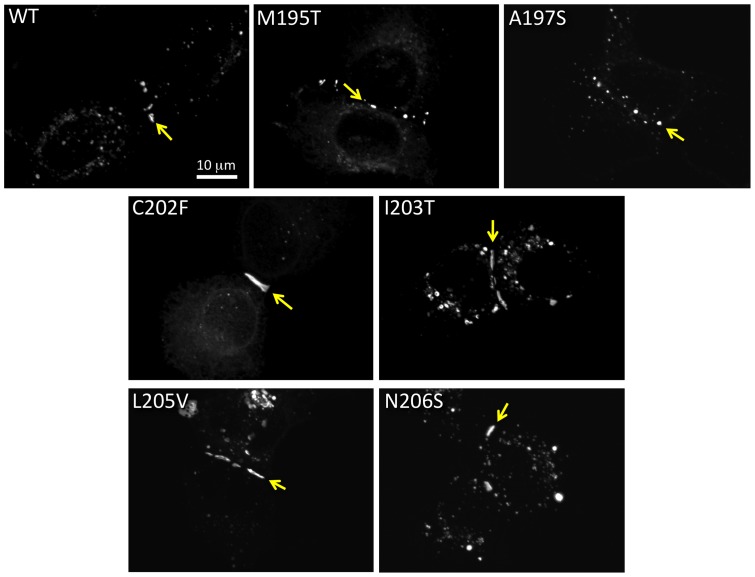
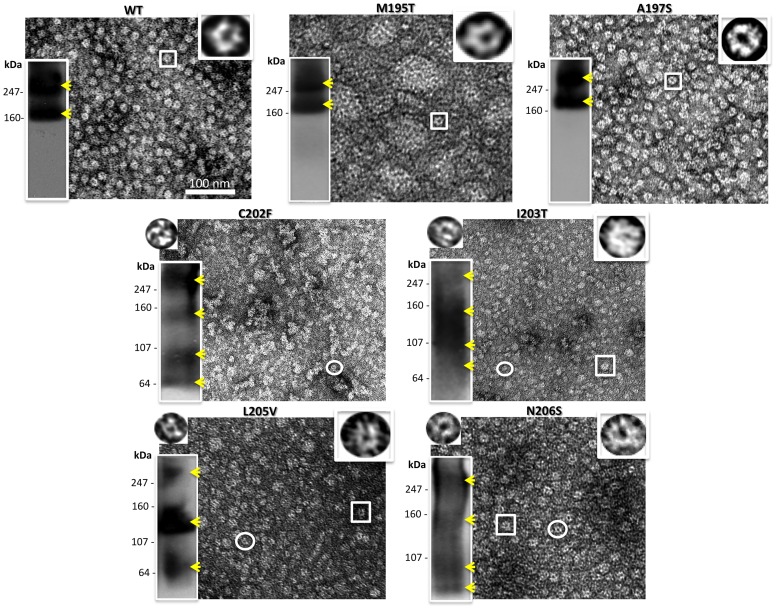
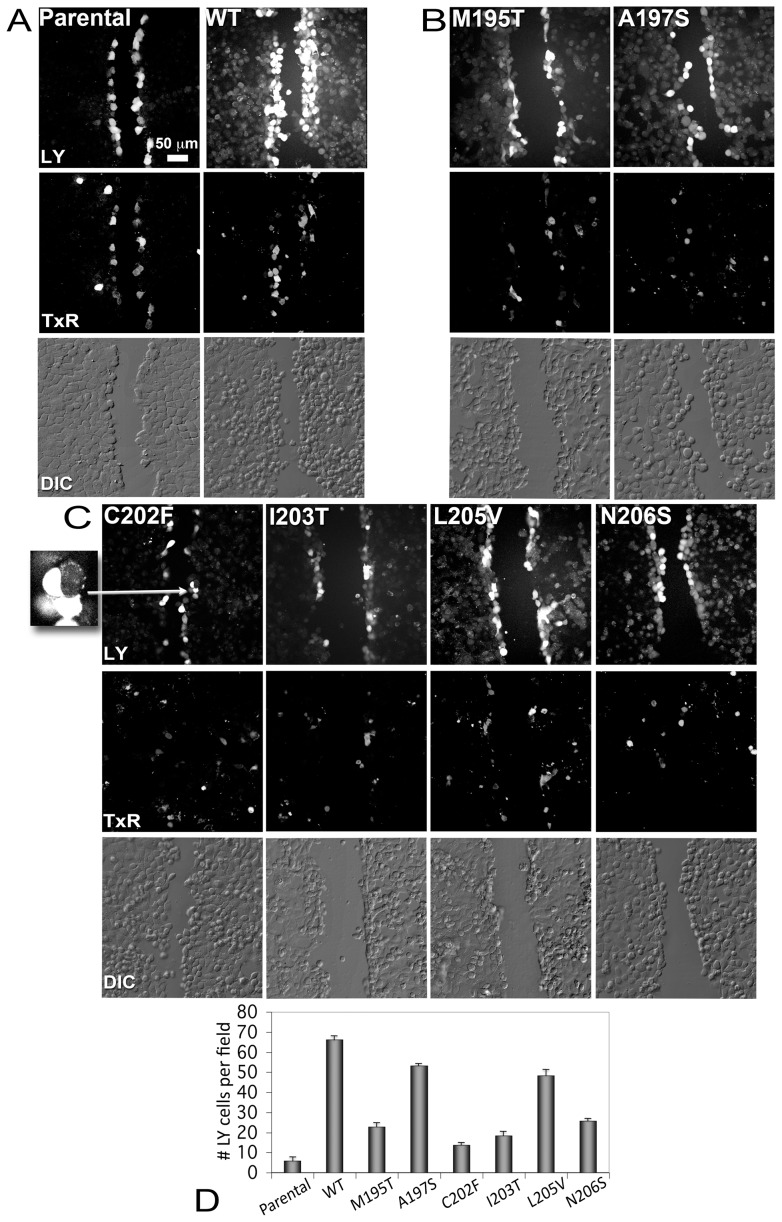
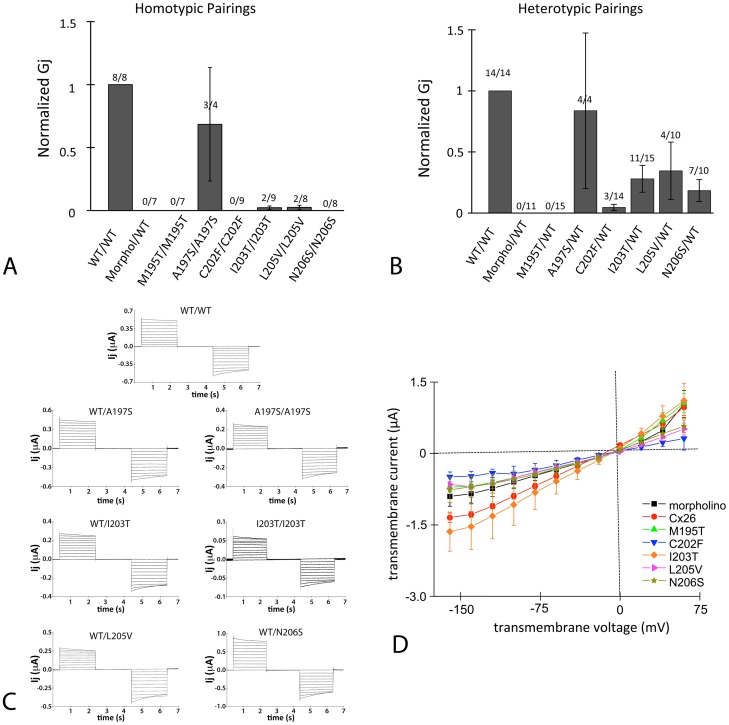
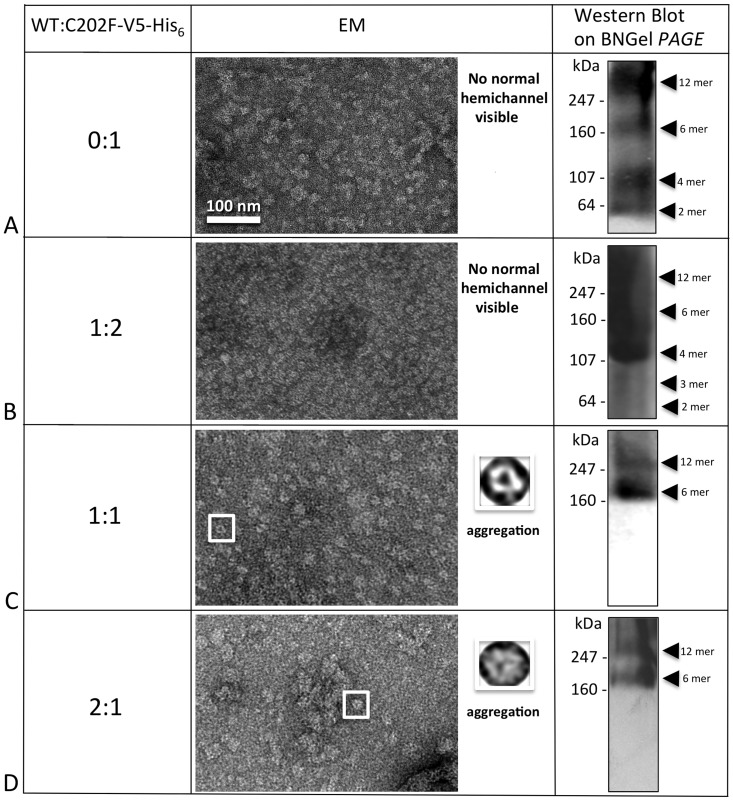
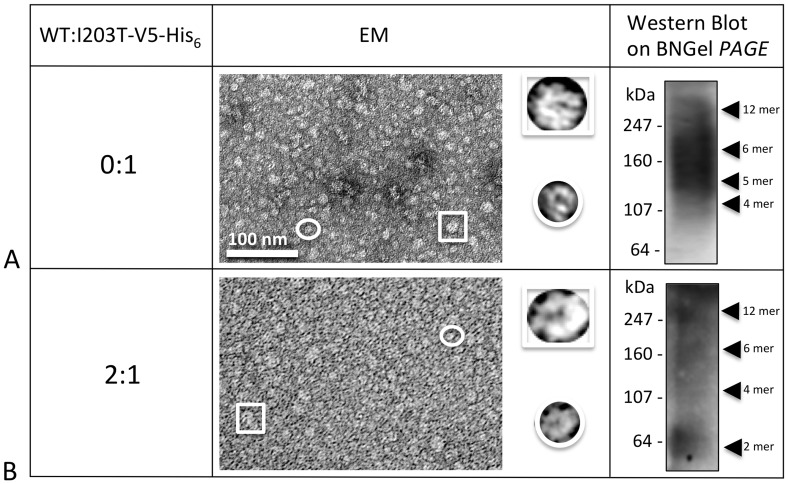
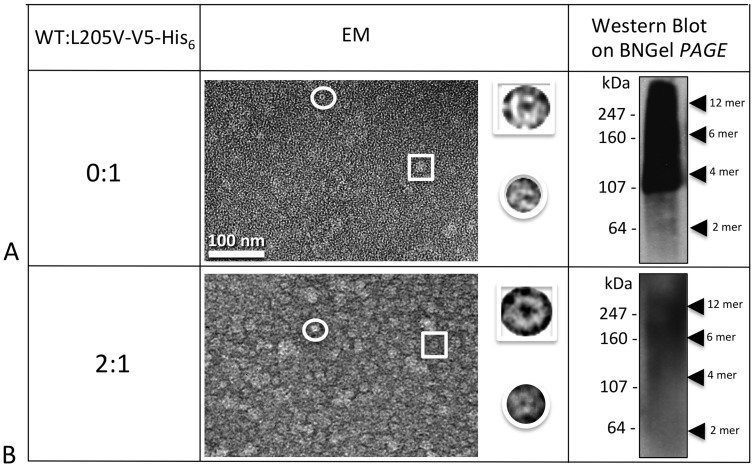
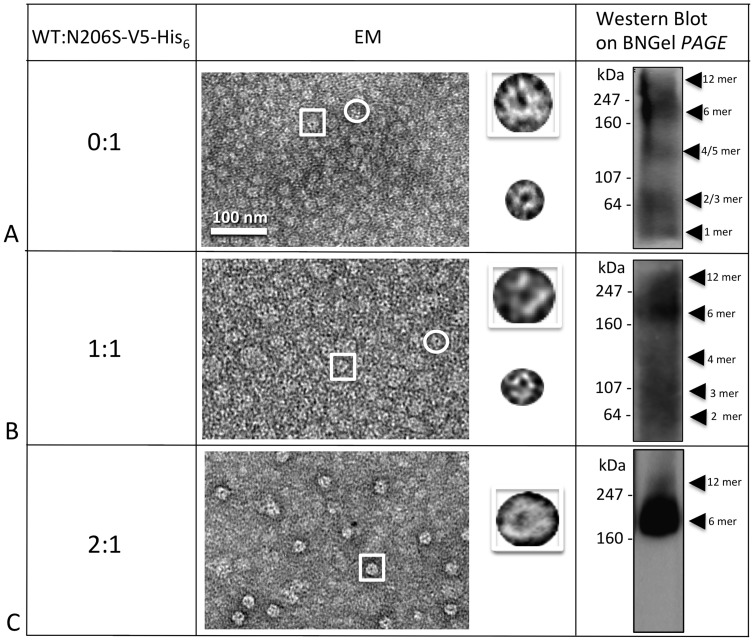
Human Connexin26 gene mutations cause hearing loss. These hereditary mutations are the leading cause of childhood deafness worldwide. Mutations in gap junction proteins (connexins) can impair intercellular communication by eliminating protein synthesis, mis-trafficking, or inducing channels that fail to dock or have aberrant function. We previously identified a new class of mutants that form non-functional gap junction channels and hemichannels (connexons) by disrupting packing and inter-helix interactions. Here we analyzed fourteen point mutations in the fourth transmembrane helix of connexin26 (Cx26) that cause non-syndromic hearing loss. Eight mutations caused mis-trafficking (K188R, F191L, V198M, S199F, G200R, I203K, L205P, T208P). Of the remaining six that formed gap junctions in mammalian cells, M195T and A197S formed stable hemichannels after isolation with a baculovirus/Sf9 protein purification system, while C202F, I203T, L205V and N206S formed hemichannels with varying degrees of instability. The function of all six gap junction-forming mutants was further assessed through measurement of dye coupling in mammalian cells and junctional conductance in paired Xenopus oocytes. Dye coupling between cell pairs was reduced by varying degrees for all six mutants. In homotypic oocyte pairings, only A197S induced measurable conductance. In heterotypic pairings with wild-type Cx26, five of the six mutants formed functional gap junction channels, albeit with reduced efficiency. None of the mutants displayed significant alterations in sensitivity to transjunctional voltage or induced conductive hemichannels in single oocytes. Intra-hemichannel interactions between mutant and wild-type proteins were assessed in rescue experiments using baculovirus expression in Sf9 insect cells. Of the four unstable mutations (C202F, I203T, L205V, N206S) only C202F and N206S formed stable hemichannels when co-expressed with wild-type Cx26. Stable M195T hemichannels displayed an increased tendency to aggregate. Thus, mutations in TM4 cause a range of phenotypes of dysfunctional gap junction channels that are discussed within the context of the X-ray crystallographic structure.
人类连接蛋白 26 基因突变导致听力损失。这些遗传性突变是全球儿童耳聋的主要原因。缝隙连接蛋白(连接蛋白)的突变可通过消除蛋白合成、错误运输或诱导无法对接或功能异常的通道来损害细胞间通讯。我们之前发现了一类新的突变体,它们通过破坏包装和螺旋间相互作用形成无功能的缝隙连接通道和半通道(连接子)。在这里,我们分析了导致非综合征性听力损失的连接蛋白 26(Cx26)第四跨膜螺旋中的 14 个点突变。8 个突变导致错误运输(K188R、F191L、V198M、S199F、G200R、I203K、L205P、T208P)。在其余在哺乳动物细胞中形成缝隙连接的 6 个突变体中,M195T 和 A197S 在使用杆状病毒/Sf9 蛋白纯化系统分离后形成稳定的半通道,而 C202F、I203T、L205V 和 N206S 则形成具有不同程度不稳定性的半通道。通过测量哺乳动物细胞中的染料偶联和配对非洲爪蟾卵母细胞中的连接电导,进一步评估了所有 6 个缝隙连接形成突变体的功能。所有 6 个突变体的细胞对之间的染料偶联均有不同程度的降低。在同种卵母细胞配对中,只有 A197S 诱导可测量的电导。在与野生型 Cx26 的异质配对中,6 个突变体中有 5 个形成功能性缝隙连接通道,尽管效率降低。在单个卵母细胞中,没有突变体显示出对跨连接电压的敏感性或诱导性导电半通道的显著改变。在 Sf9 昆虫细胞中使用杆状病毒表达进行挽救实验,评估了突变体和野生型蛋白之间的半通道内相互作用。在不稳定的 4 个突变体(C202F、I203T、L205V、N206S)中,只有 C202F 和 N206S 与野生型 Cx26 共表达时形成稳定的半通道。稳定的 M195T 半通道显示出聚集的趋势增加。因此,TM4 中的突变导致功能失调的缝隙连接通道的一系列表型,这些表型在 X 射线晶体结构的背景下进行了讨论。