Department of Medical Genetics, College of Medicine, Umm Al-Qura University, Mecca, 21955, Saudi Arabia.
Department of Community Medicine, College of Medicine, Umm Al-Qura University, Mecca, Saudi Arabia.
Hum Genomics. 2023 Jul 7;17(1):60. doi: 10.1186/s40246-023-00507-2.
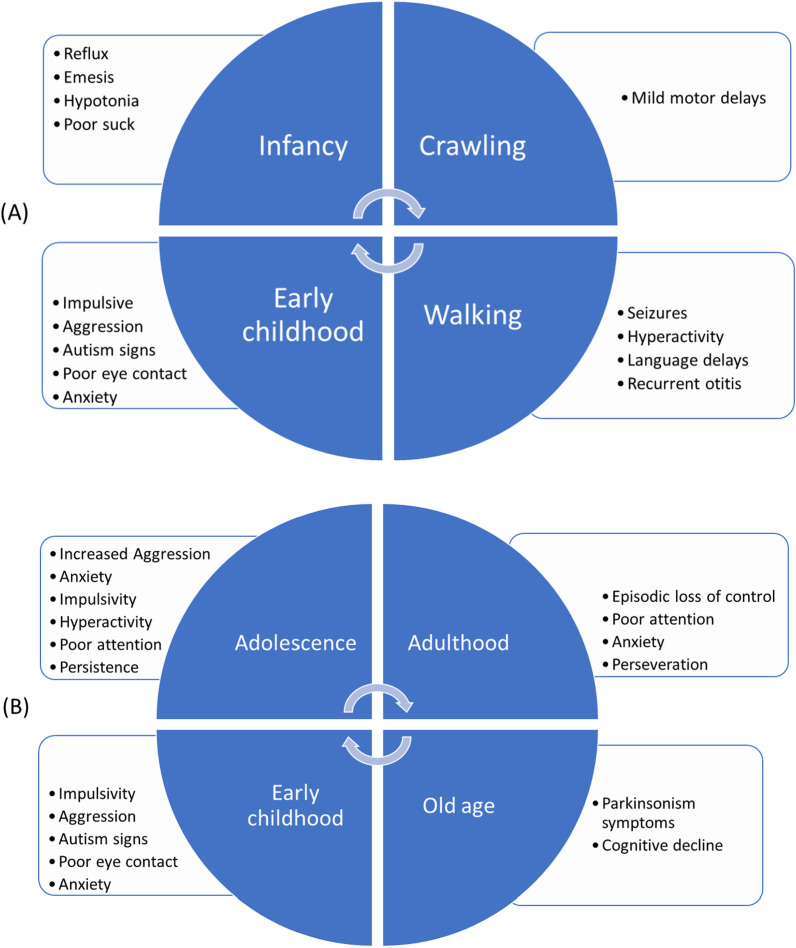
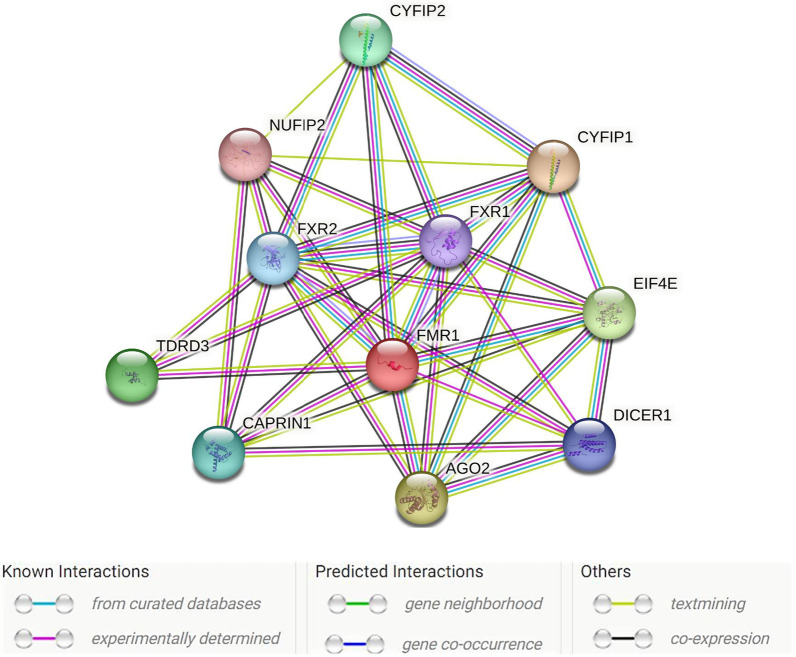
This review discusses the discovery, epidemiology, pathophysiology, genetic etiology, molecular diagnosis, and medication-based management of fragile X syndrome (FXS). It also highlights the syndrome's variable expressivity and common comorbid and overlapping conditions. FXS is an X-linked dominant disorder associated with a wide spectrum of clinical features, including but not limited to intellectual disability, autism spectrum disorder, language deficits, macroorchidism, seizures, and anxiety. Its prevalence in the general population is approximately 1 in 5000-7000 men and 1 in 4000-6000 women worldwide. FXS is associated with the fragile X messenger ribonucleoprotein 1 (FMR1) gene located at locus Xq27.3 and encodes the fragile X messenger ribonucleoprotein (FMRP). Most individuals with FXS have an FMR1 allele with > 200 CGG repeats (full mutation) and hypermethylation of the CpG island proximal to the repeats, which silences the gene's promoter. Some individuals have mosaicism in the size of the CGG repeats or in hypermethylation of the CpG island, both produce some FMRP and give rise to milder cognitive and behavioral deficits than in non-mosaic individuals with FXS. As in several monogenic disorders, modifier genes influence the penetrance of FMR1 mutations and FXS's variable expressivity by regulating the pathophysiological mechanisms related to the syndrome's behavioral features. Although there is no cure for FXS, prenatal molecular diagnostic testing is recommended to facilitate early diagnosis. Pharmacologic agents can reduce some behavioral features of FXS, and researchers are investigating whether gene editing can be used to demethylate the FMR1 promoter region to improve patient outcomes. Moreover, clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 and developed nuclease defective Cas9 (dCas9) strategies have promised options of genome editing in gain-of-function mutations to rewrite new genetic information into a specified DNA site, are also being studied.
本文综述了脆性 X 综合征(FXS)的发现、流行病学、病理生理学、遗传病因学、分子诊断和基于药物的治疗管理。本文还强调了该综合征的可变表达性以及常见的合并症和重叠病症。FXS 是一种 X 连锁显性遗传疾病,与广泛的临床特征相关,包括但不限于智力障碍、自闭症谱系障碍、语言缺陷、巨睾症、癫痫发作和焦虑。其在全球普通人群中的患病率约为每 5000-7000 名男性和每 4000-6000 名女性中存在 1 例。FXS 与位于 Xq27.3 基因座的脆性 X 信使核糖核蛋白 1(FMR1)基因相关,该基因编码脆性 X 信使核糖核蛋白(FMRP)。大多数 FXS 患者的 FMR1 等位基因有超过 200 个 CGG 重复(完全突变),并且重复序列近端的 CpG 岛超甲基化,从而使基因启动子沉默。一些个体的 CGG 重复序列大小或 CpG 岛的超甲基化呈镶嵌状态,这两种情况都会产生一些 FMRP,并导致比 FXS 非镶嵌个体更轻微的认知和行为缺陷。与几种单基因疾病一样,修饰基因通过调节与该综合征行为特征相关的病理生理机制,影响 FMR1 突变的外显率和 FXS 的可变表达性。尽管 FXS 尚无治愈方法,但推荐进行产前分子诊断测试以促进早期诊断。药物治疗可减轻 FXS 的某些行为特征,研究人员正在研究是否可以使用基因编辑来使 FMR1 启动子区域去甲基化,以改善患者的治疗效果。此外,成簇规律间隔短回文重复(CRISPR)/Cas9 和开发的核酸酶缺陷 Cas9(dCas9)策略有望用于功能获得性突变的基因组编辑,将新的遗传信息重写到指定的 DNA 位点,这些策略也正在研究中。