Martinez-Carrasco Alejandro, Real Raquel, Lawton Michael, Iwaki Hirotaka, Tan Manuela M X, Wu Lesley, Williams Nigel M, Carroll Camille, Hu Michele T M, Grosset Donald G, Hardy John, Ryten Mina, Foltynie Tom, Ben-Shlomo Yoav, Shoai Maryam, Morris Huw R
Department of Clinical and Movement Neurosciences, UCL Queen Square Institute of Neurology, University College London, UK.
UCL Movement Disorders Centre, University College London, London, UK.
medRxiv. 2023 May 30:2023.05.24.23290362. doi: 10.1101/2023.05.24.23290362.
Forty percent of Parkinson's disease patients develop levodopa-induced-dyskinesia (LiD) within 4 years of starting levodopa. The genetic basis of LiD remains poorly understood, and there have been few well powered studies.
To discover common genetic variants in the PD population that increase the probability of developing LiD.
We performed survival analyses to study the development of LiD in 5 separate longitudinal cohorts. We performed a meta-analysis to combine the results of genetic association from each study based on a fixed effects model weighting the effect sizes by the inverse of their standard error. The selection criteria was specific to each cohort. We studied individuals that were genotyped from each cohort and that passed our analysis specific inclusion criteria.
We measured the time for PD patients on levodopa treatment to develop LiD as defined by reaching a score higher or equal than 2 from the MDS-UPDRS part IV, item 1, which is equivalent to a range of 26%-50% of the waking time with dyskinesia. We carried out a genome-wide analysis of the hazard ratio and the association of genome-wide SNPs with the probability of developing LiD using cox proportional hazard models (CPH).
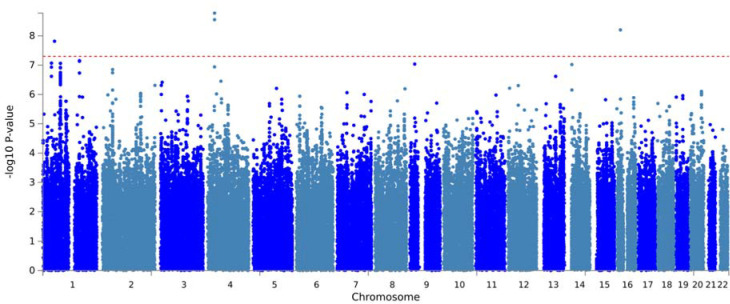
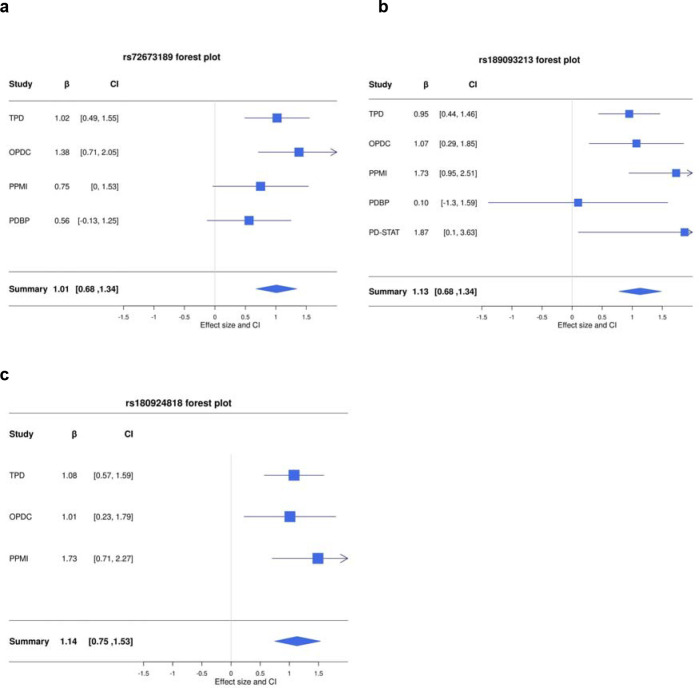
This study included 2,784 PD patients of European ancestry, of whom 14.6% developed LiD. Consistent with previous studies, we found female gender (HR = 1.35, SE = 0.11, = 0.007) and younger age at onset (HR = 1.8, SE = 0.14, = 2 × 10 ) to increase the probability of developing LiD. We identified three loci significantly associated with time-to-LiD onset. on chromosome 1 (HR = 2.77, SE = 0.18, = 1.53 × 10 ) located in the LRP8 locus, chromosome 4 (HR = 3.06,, SE = 0.19, = 2.81 × 10 ) in the non-coding RNA locus, and on chromosome 16 (HR = 3.13, SE = 0.20, = 6.27 × 10 ) in the locus. Subsequent colocalization analyses on chromosome 1 identified as a candidate gene associated with LiD through a change in gene expression. We computed a PRS based on our GWAS meta-analysis and found high accuracy to stratify between PD-LID and PD (AUC 83.9). We also performed a stepwise regression analysis for baseline features selection associated with LiD status. We found baseline anxiety status to be significantly associated with LiD (OR = 1.14, SE = 0.03, = 7.4 × 10 ). Finally, we performed a candidate variant analysis and found that genetic variability in ( , Beta = 0.24, SE = 0.09, = 8.89 × 10 ) and ( , Beta = 0.19, SE = 0.10, = 4.95 × 10 ) loci were significantly associated with time to LiD in our large meta-analysis.
In this association study, we have found three novel genetic variants associated with LiD, as well as confirming reports that variability in ANKK1 and BDNF loci were significantly associated with LiD probability. A PRS nominated from our time-to-LiD meta-analysis significantly differentiated between PD-LiD and PD. In addition, we have found female gender, young PD onset and anxiety to be significantly associated with LiD.
40%的帕金森病患者在开始使用左旋多巴治疗4年内会出现左旋多巴诱导的异动症(LiD)。LiD的遗传基础仍知之甚少,且鲜有大规模的研究。
在帕金森病患者群体中发现增加发生LiD可能性的常见基因变异。
设计、背景与参与者:我们进行了生存分析,以研究5个独立纵向队列中LiD的发生情况。我们基于固定效应模型进行荟萃分析,将每项研究的基因关联结果合并,权重为效应量的标准误的倒数。每个队列的选择标准各不相同。我们研究了每个队列中经过基因分型且符合我们分析特定纳入标准的个体。
我们测量了接受左旋多巴治疗的帕金森病患者出现LiD的时间,其定义为从MDS-UPDRS第四部分第1项达到或高于2分,这相当于异动症占清醒时间的26%-50%。我们使用Cox比例风险模型(CPH)对风险比以及全基因组单核苷酸多态性(SNP)与发生LiD的概率进行了全基因组分析。
本研究纳入了2784名欧洲血统的帕金森病患者,其中14.6%出现了LiD。与先前的研究一致,我们发现女性(风险比=1.35,标准误=0.11,P=0.007)和发病年龄较轻(风险比=1.8,标准误=0.14,P=2×10⁻⁵)会增加发生LiD的可能性。我们确定了三个与LiD发病时间显著相关的基因座。位于染色体1上的LRP8基因座(风险比=2.77,标准误=0.18,P=1.53×10⁻⁷),位于染色体4上非编码RNA基因座(风险比=3.06,标准误=0.19,P=2.81×10⁻⁸),以及位于染色体16上的基因座(风险比=3.13,标准误=0.20,P=6.27×10⁻⁸)。随后在染色体1上进行的共定位分析确定,通过基因表达的变化,该基因座是与LiD相关的候选基因。我们基于全基因组关联研究(GWAS)荟萃分析计算了多基因风险评分(PRS),发现其在区分帕金森病合并LiD(PD-LID)和帕金森病(PD)方面具有较高的准确性(曲线下面积[AUC]为83.9)。我们还对与LiD状态相关的基线特征进行了逐步回归分析。我们发现基线焦虑状态与LiD显著相关(比值比=1.14,标准误=0.03,P=7.4×10⁻⁵)。最后,我们进行了候选变异分析,发现在我们的大型荟萃分析中,基因座和基因座的遗传变异性与LiD发生时间显著相关(,β=0.24,标准误=0.09,P=8.89×10⁻³)(,β=0.19,标准误=0.10,P=4.95×10⁻²)。
在这项关联研究中,我们发现了三个与LiD相关的新基因变异,同时也证实了ANKK1和BDNF基因座的变异性与LiD发生概率显著相关的报道。我们从LiD发病时间的荟萃分析中得出的PRS能够显著区分PD-LID和PD。此外,我们发现女性、帕金森病发病年龄较轻和焦虑与LiD显著相关。