Lassen Frederik H, Venkatesh Samvida S, Baya Nikolas, Zhou Wei, Bloemendal Alex, Neale Benjamin M, Kessler Benedikt M, Whiffin Nicola, Lindgren Cecilia M, Palmer Duncan S
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom.
Big Data Institute, Li Ka Shing Centre for Health Information and Discovery, University of Oxford, Oxford, United Kingdom.
medRxiv. 2023 Jul 3:2023.06.29.23291992. doi: 10.1101/2023.06.29.23291992.
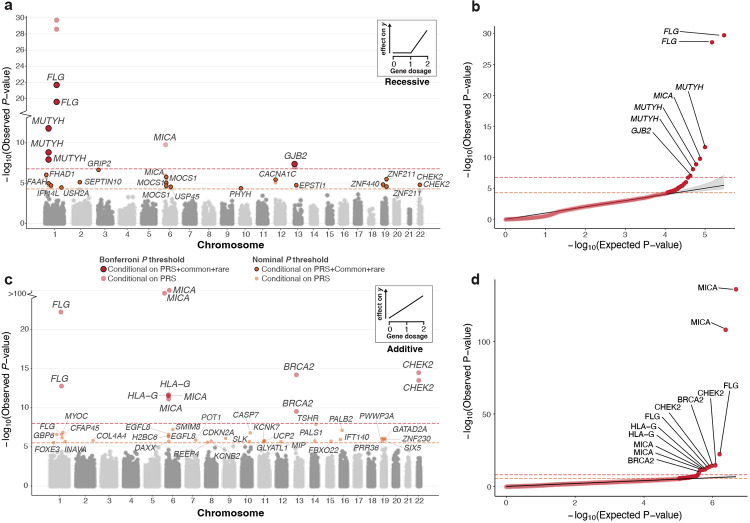
Exome-sequencing association studies have successfully linked rare protein-coding variation to risk of thousands of diseases. However, the relationship between rare deleterious compound heterozygous (CH) variation and their phenotypic impact has not been fully investigated. Here, we leverage advances in statistical phasing to accurately phase rare variants (MAF ~ 0.001%) in exome sequencing data from 175,587 UK Biobank (UKBB) participants, which we then systematically annotate to identify putatively deleterious CH coding variation. We show that 6.5% of individuals carry such damaging variants in the CH state, with 90% of variants occurring at MAF < 0.34%. Using a logistic mixed model framework, systematically accounting for relatedness, polygenic risk, nearby common variants, and rare variant burden, we investigate recessive effects in common complex diseases. We find six exome-wide significant () and 17 nominally significant () gene-trait associations. Among these, only four would have been identified without accounting for CH variation in the gene. We further incorporate age-at-diagnosis information from primary care electronic health records, to show that genetic phase influences lifetime risk of disease across 20 gene-trait combinations (FDR < 5%). Using a permutation approach, we find evidence for genetic phase contributing to disease susceptibility for a collection of gene-trait pairs, including -asthma () and -visual impairment (). Taken together, we demonstrate the utility of phasing large-scale genetic sequencing cohorts for robust identification of the phenome-wide consequences of compound heterozygosity.
外显子组测序关联研究已成功地将罕见的蛋白质编码变异与数千种疾病的风险联系起来。然而,罕见有害复合杂合(CH)变异与其表型影响之间的关系尚未得到充分研究。在此,我们利用统计定相技术的进展,对来自175587名英国生物银行(UKBB)参与者的外显子组测序数据中的罕见变异(MAF约0.001%)进行准确定相,然后我们对其进行系统注释,以识别可能有害的CH编码变异。我们发现6.5%的个体携带处于CH状态的此类有害变异,其中90%的变异发生在MAF<0.34%时。使用逻辑混合模型框架,系统地考虑亲缘关系、多基因风险、附近的常见变异和罕见变异负担,我们研究常见复杂疾病中的隐性效应。我们发现了6个全外显子组显著()和17个名义上显著()的基因-性状关联。其中,只有4个在不考虑基因中的CH变异时才能被识别出来。我们进一步纳入来自初级保健电子健康记录的诊断年龄信息,以表明遗传定相影响20种基因-性状组合的终身疾病风险(FDR<5%)。使用排列方法,我们发现遗传定相有助于一系列基因-性状对的疾病易感性的证据,包括-哮喘()和-视力损害()。综上所述,我们证明了对大规模基因测序队列进行定相对于可靠识别复合杂合性在全表型范围内的后果的实用性。