Institute of Anatomy and Cell Biology, Ulm University, Ulm, Germany.
Division of Cellular and Systems Medicine, School of Medicine, University of Dundee, Dundee, Scotland, UK.
Acta Neuropathol. 2023 Sep;146(3):451-475. doi: 10.1007/s00401-023-02611-y. Epub 2023 Jul 24.
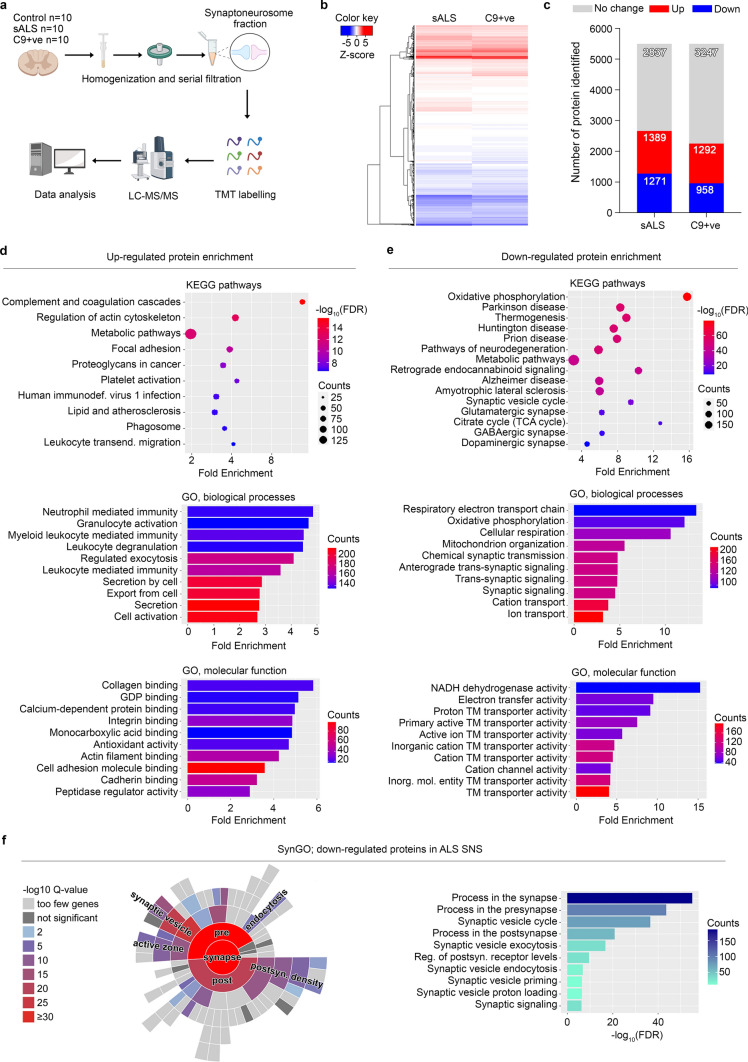
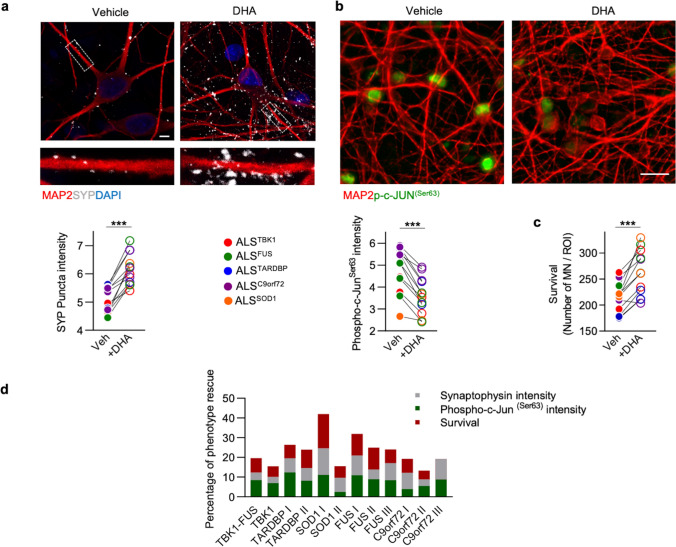
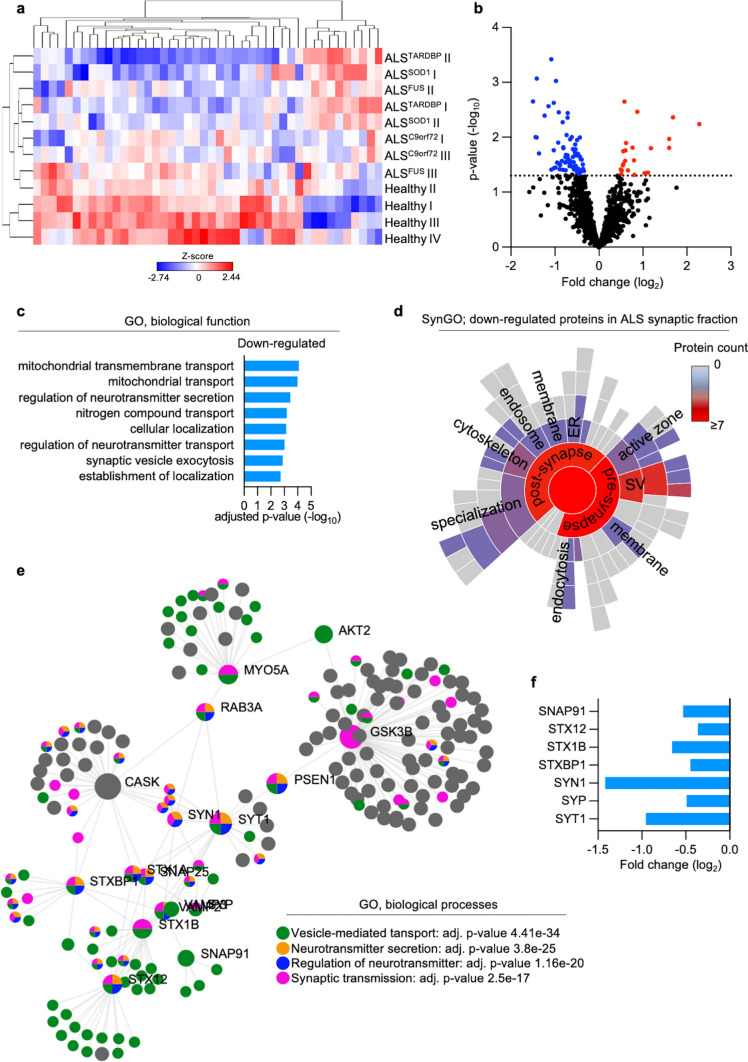
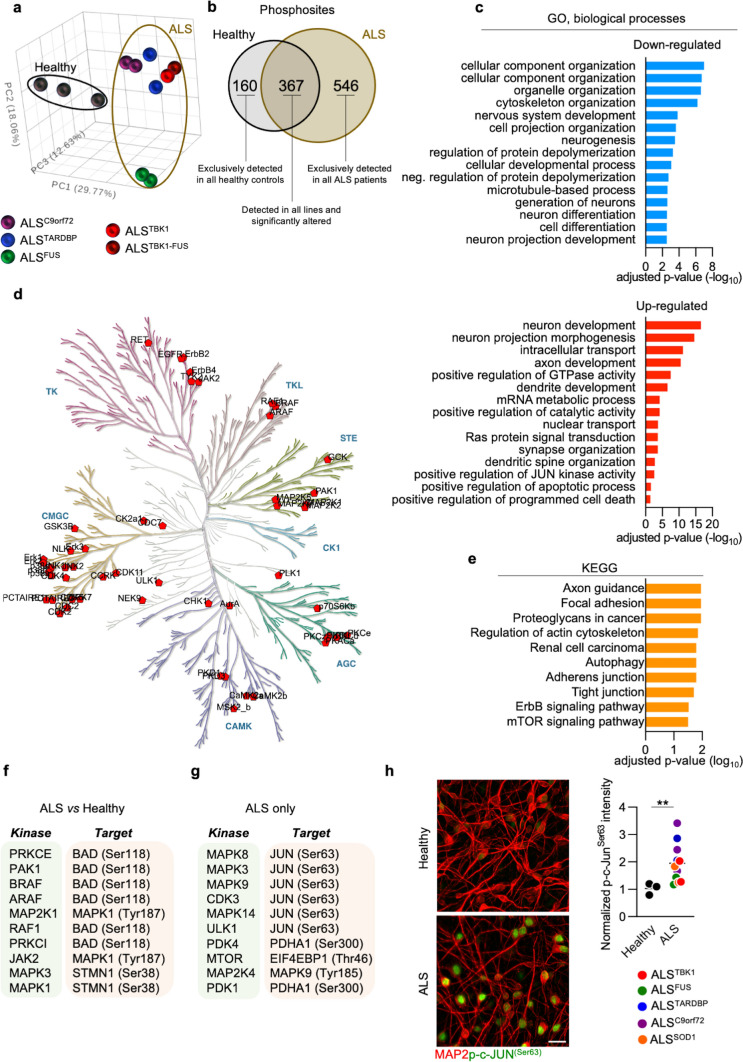
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease mainly affecting upper and lower motoneurons. Several functionally heterogeneous genes have been associated with the familial form of this disorder (fALS), depicting an extremely complex pathogenic landscape. This heterogeneity has limited the identification of an effective therapy, and this bleak prognosis will only improve with a greater understanding of convergent disease mechanisms. Recent evidence from human post-mortem material and diverse model systems has highlighted the synapse as a crucial structure actively involved in disease progression, suggesting that synaptic aberrations might represent a shared pathological feature across the ALS spectrum. To test this hypothesis, we performed the first comprehensive analysis of the synaptic proteome from post-mortem spinal cord and human iPSC-derived motoneurons carrying mutations in the major ALS genes. This integrated approach highlighted perturbations in the molecular machinery controlling vesicle release as a shared pathomechanism in ALS. Mechanistically, phosphoproteomic analysis linked the presynaptic vesicular phenotype to an accumulation of cytotoxic protein aggregates and to the pro-apoptotic activation of the transcription factor c-Jun, providing detailed insights into the shared pathobiochemistry in ALS. Notably, sub-chronic treatment of our iPSC-derived motoneurons with the fatty acid docosahexaenoic acid exerted a neuroprotective effect by efficiently rescuing the alterations revealed by our multidisciplinary approach. Together, this study provides strong evidence for the central and convergent role played by the synaptic microenvironment within the ALS spinal cord and highlights a potential therapeutic target that counteracts degeneration in a heterogeneous cohort of human motoneuron cultures.
肌萎缩侧索硬化症(ALS)是一种致命的神经退行性疾病,主要影响上下运动神经元。几种功能不同的基因已与该疾病的家族形式(fALS)相关联,描绘了极其复杂的发病机制。这种异质性限制了有效治疗方法的确定,只有对趋同疾病机制有更深入的了解,才能改善这一黯淡的预后。最近来自人类死后组织和多种模型系统的证据强调了突触作为一个关键结构,积极参与疾病进展,表明突触异常可能是 ALS 谱中共同的病理特征。为了验证这一假设,我们对来自死后脊髓和携带主要 ALS 基因突变的人诱导多能干细胞衍生运动神经元的突触蛋白质组进行了首次全面分析。这种综合方法突出了控制囊泡释放的分子机制的扰动是 ALS 的共同病理机制。从机制上讲,磷酸蛋白质组学分析将突触小泡表型与细胞毒性蛋白聚集体的积累以及转录因子 c-Jun 的促凋亡激活联系起来,为 ALS 中的共同病理生物化学提供了详细的见解。值得注意的是,我们用多学科方法揭示的改变,我们用脂肪酸二十二碳六烯酸对源自 iPSC 的运动神经元进行亚慢性处理,发挥了神经保护作用,有效地挽救了这些改变。总之,这项研究为 ALS 脊髓中的突触微环境的核心和趋同作用提供了有力的证据,并突出了一个潜在的治疗靶点,该靶点可以在异质的人类运动神经元培养物中对抗退化。