Azuero Oscar Cortés, Lefrancq Noémie, Nikolay Birgit, McKee Clifton, Cappelle Julien, Hul Vibol, Ou Tey Putita, Hoem Thavry, Lemey Philippe, Rahman Mohammed Ziaur, Islam Ausraful, Gurley Emily S, Duong Veasna, Salje Henrik
Department of Genetics, University of Cambridge, Cambridge CB2 3EH, UK.
Epicentre, 75019 Paris, France.
medRxiv. 2023 Oct 4:2023.07.14.23292668. doi: 10.1101/2023.07.14.23292668.
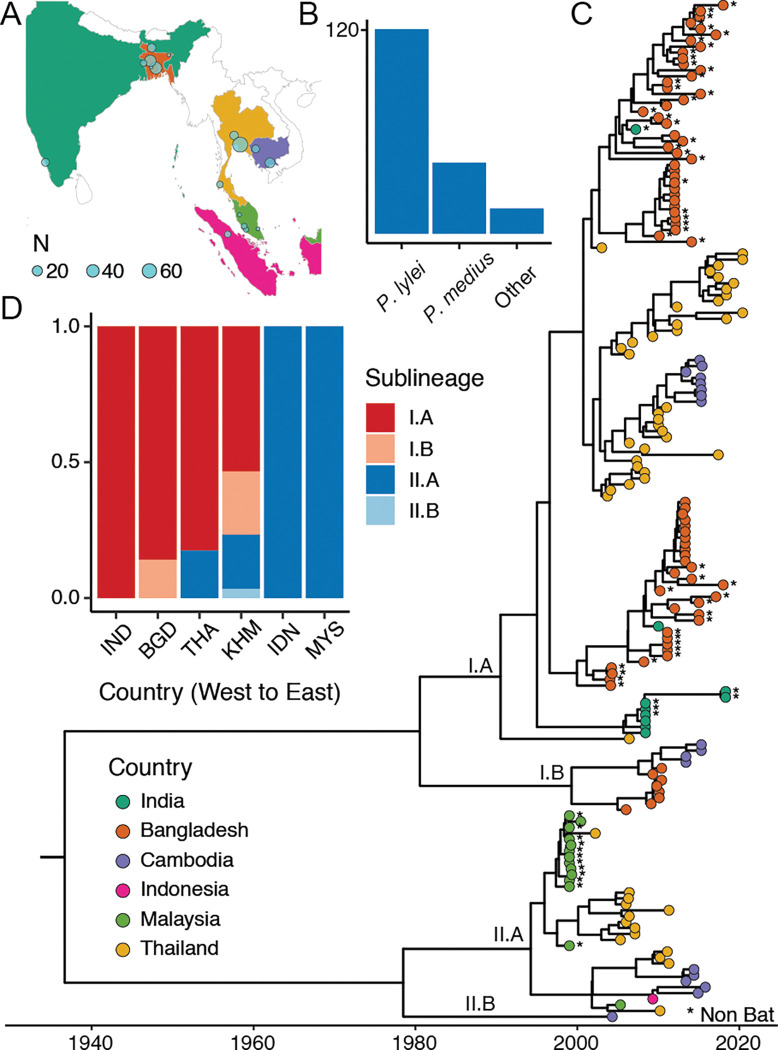
Nipah virus (NiV), a highly lethal virus in humans, circulates silently in bats throughout South and Southeast Asia. Difficulty in obtaining genomes from bats means we have a poor understanding of NiV diversity, including how many lineages circulate within a roost and the spread of NiV over increasing spatial scales. Here we develop phylogenetic approaches applied to the most comprehensive collection of genomes to date (N=257, 175 from bats, 73 from humans) from six countries over 22 years (1999-2020). In Bangladesh, where most human infections occur, we find evidence of increased spillover risk from one of the two co-circulating sublineages. We divide the four major NiV sublineages into 15 genetic clusters (emerged 20-44 years ago). Within any bat roost, there are an average of 2.4 co-circulating genetic clusters, rising to 5.5 clusters at areas of 1,500-2,000 km. Using Approximate Bayesian Computation fit to a spatial signature of viral diversity, we estimate that each genetic cluster occupies an average area of 1.3 million km (95%CI: 0.6-2.3 million), with 14 clusters in an area of 100,000 km (95%CI: 6-24). In the few sites in Bangladesh and Cambodia where genomic surveillance has been concentrated, we estimate that most of the genetic clusters have been identified, but only ~15% of overall NiV diversity has been uncovered. Our findings are consistent with entrenched co-circulation of distinct lineages, even within individual roosts, coupled with slow migration over larger spatial scales.
尼帕病毒(NiV)是一种对人类具有高度致死性的病毒,在南亚和东南亚的蝙蝠中悄然传播。从蝙蝠身上获取基因组存在困难,这意味着我们对尼帕病毒的多样性了解不足,包括在一个栖息地内有多少谱系在传播以及尼帕病毒在不断扩大的空间尺度上的传播情况。在此,我们开发了系统发育方法,应用于来自六个国家22年(1999 - 2020年)以来最全面的基因组集合(N = 257,其中175个来自蝙蝠,73个来自人类)。在大多数人类感染发生的孟加拉国,我们发现了来自两个共同传播的亚谱系之一的溢出风险增加的证据。我们将四个主要的尼帕病毒亚谱系分为15个遗传簇(在20 - 44年前出现)。在任何蝙蝠栖息地内,平均有2.4个共同传播的遗传簇,在1500 - 2000公里的区域内增加到5.5个簇。使用拟合病毒多样性空间特征的近似贝叶斯计算方法,我们估计每个遗传簇平均占据130万平方公里的面积(95%可信区间:60万 - 230万),在10万平方公里的区域内有14个簇(95%可信区间:6万 - 24万)。在孟加拉国和柬埔寨少数集中进行基因组监测的地点,我们估计大多数遗传簇已被识别,但仅发现了约15%的尼帕病毒总体多样性。我们的研究结果与不同谱系的长期共同传播一致,即使在单个栖息地内也是如此,同时在更大的空间尺度上迁移缓慢。