Rossino Giacomo, Marra Annamaria, Listro Roberta, Peviani Marco, Poggio Elena, Curti Daniela, Pellavio Giorgia, Laforenza Umberto, Dondio Giulio, Schepmann Dirk, Wünsch Bernhard, Bedeschi Martina, Marino Noemi, Tesei Anna, Ha Hee-Jin, Kim Young-Ho, Ann Jihyae, Lee Jeewoo, Linciano Pasquale, Di Giacomo Marcello, Rossi Daniela, Collina Simona
Department of Drug Sciences, University of Pavia, 27100 Pavia, Italy.
Department of Biology and Biotechnology "L. Spallanzani", University of Pavia, 27100 Pavia, Italy.
Pharmaceuticals (Basel). 2023 Jul 5;16(7):962. doi: 10.3390/ph16070962.
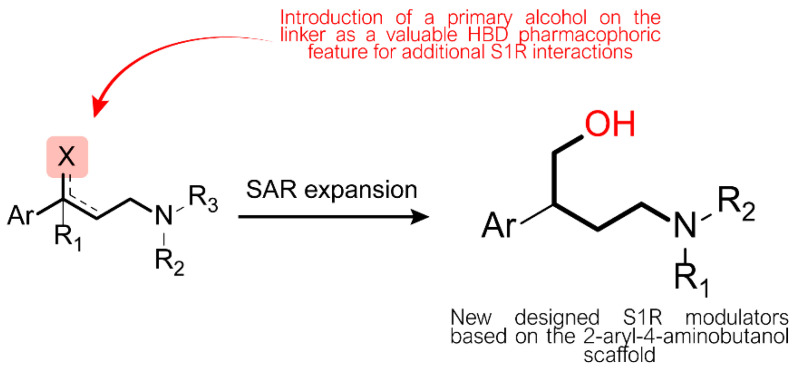
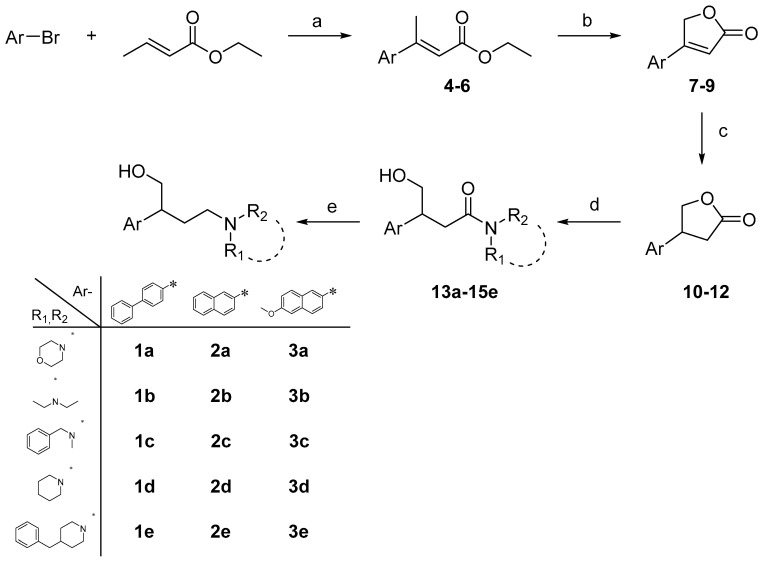
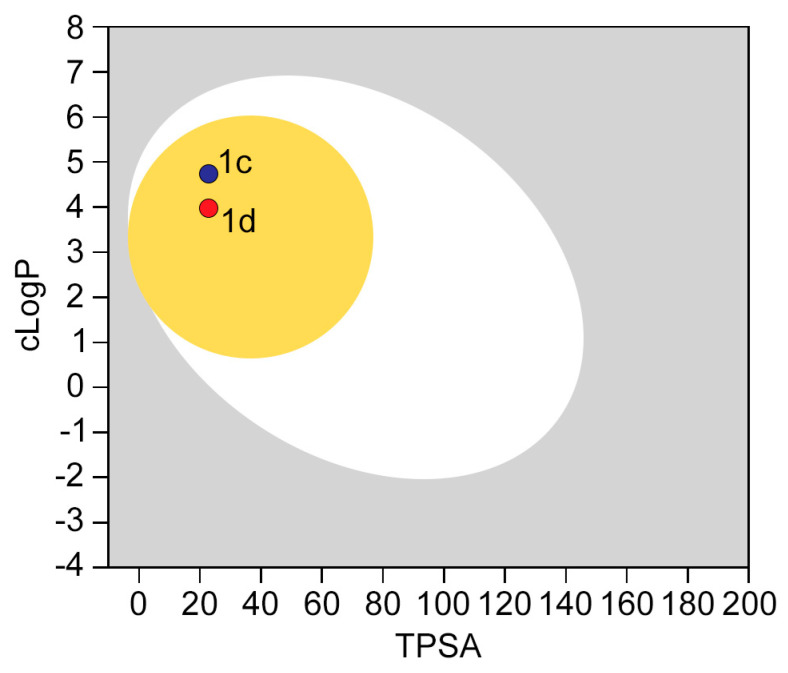
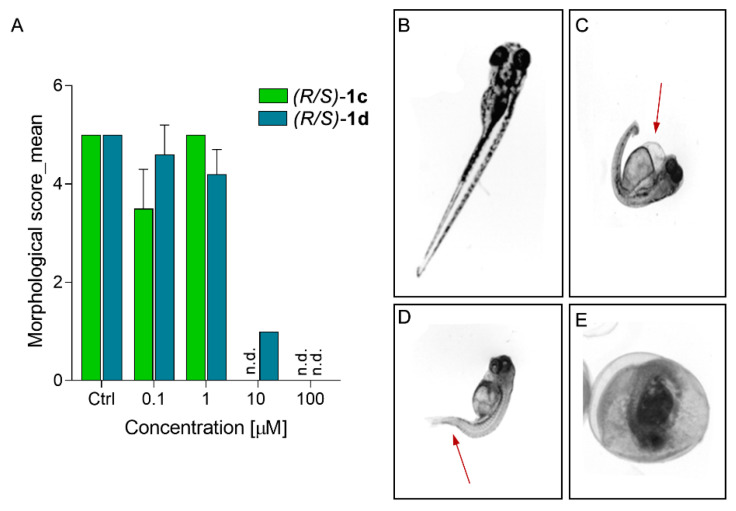
Neuropathic pain (NP) is a chronic condition resulting from damaged pain-signaling pathways. It is a debilitating disorder that affects up to 10% of the world's population. Although opioid analgesics are effective in reducing pain, they present severe risks; so, there is a pressing need for non-opioid pain-relieving drugs. One potential alternative is represented by sigma-1 receptor (S1R) antagonists due to their promising analgesic effects. Here, we report the synthesis and biological evaluation of a series of S1R antagonists based on a 2-aryl-4-aminobutanol scaffold. After assessing affinity toward the S1R and selectivity over the sigma-2 receptor (S2R), we evaluated the agonist/antagonist profile of the compounds by investigating their effects on nerve growth factor-induced neurite outgrowth and aquaporin-mediated water permeability in the presence and absence of oxidative stress. (/)-- emerged as the most interesting compound for S1R affinity ( S1R = 6.2 ± 0.9) and functional antagonist activity. Furthermore, it showed no cytotoxic effect in two normal human cell lines or in an in vivo zebrafish model and was stable after incubation in mouse plasma. (/)-- was then evaluated in two animal models of NP: the formalin test and the spinal nerve ligation model. The results clearly demonstrated that compound (/)-- effectively alleviated pain in both animal models, thus providing the proof of concept of its efficacy as an antinociceptive agent.
神经性疼痛(NP)是一种由受损的疼痛信号通路引起的慢性疾病。它是一种使人衰弱的病症,影响着全球多达10%的人口。尽管阿片类镇痛药在减轻疼痛方面有效,但它们存在严重风险;因此,迫切需要非阿片类止痛药物。由于其有前景的镇痛作用,σ-1受体(S1R)拮抗剂是一种潜在的替代药物。在此,我们报告了一系列基于2-芳基-4-氨基丁醇支架的S1R拮抗剂的合成及生物学评价。在评估了对S1R的亲和力以及对σ-2受体(S2R)的选择性后,我们通过研究化合物在有和没有氧化应激的情况下对神经生长因子诱导的神经突生长和水通道蛋白介导的水通透性的影响,来评估这些化合物的激动剂/拮抗剂特性。(/)——因其对S1R的亲和力(S = 6.2 ± 0.9)和功能性拮抗剂活性而成为最具吸引力的化合物。此外,它在两种正常人类细胞系或体内斑马鱼模型中均未显示出细胞毒性作用,并且在小鼠血浆中孵育后很稳定。(/)——随后在两种NP动物模型中进行了评估:福尔马林试验和脊神经结扎模型。结果清楚地表明,化合物(/)——在两种动物模型中均有效减轻了疼痛
,从而为其作为一种抗伤害感受剂的功效提供了概念验证。