Petrone Joseph R, Rios Glusberger Paula, George Christian D, Milletich Patricia L, Ahrens Angelica P, Roesch Luiz Fernando Wurdig, Triplett Eric W
Department of Microbiology and Cell Science, Institute of Food and Agricultural Sciences, University of Florida, Gainesville, FL, United States.
Front Microbiol. 2023 Jul 20;14:1201064. doi: 10.3389/fmicb.2023.1201064. eCollection 2023.
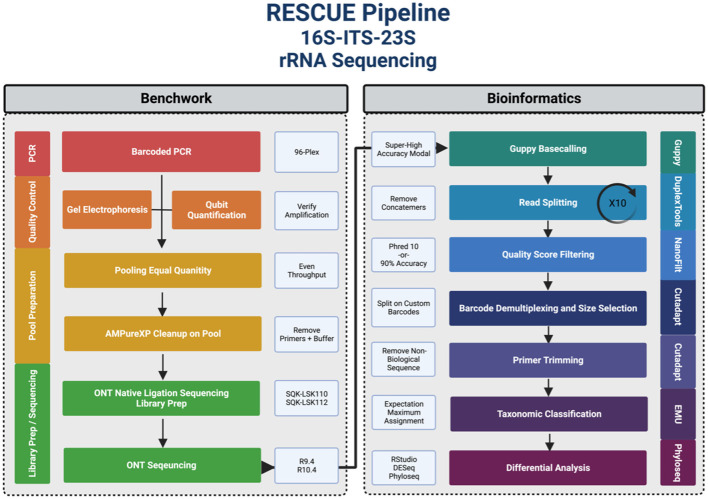
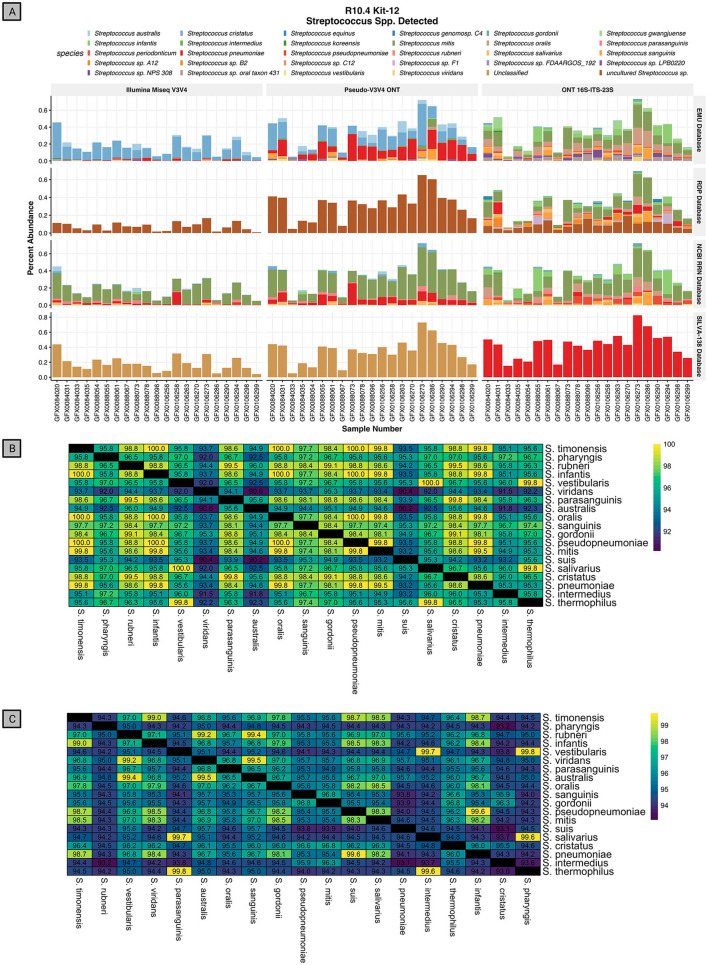
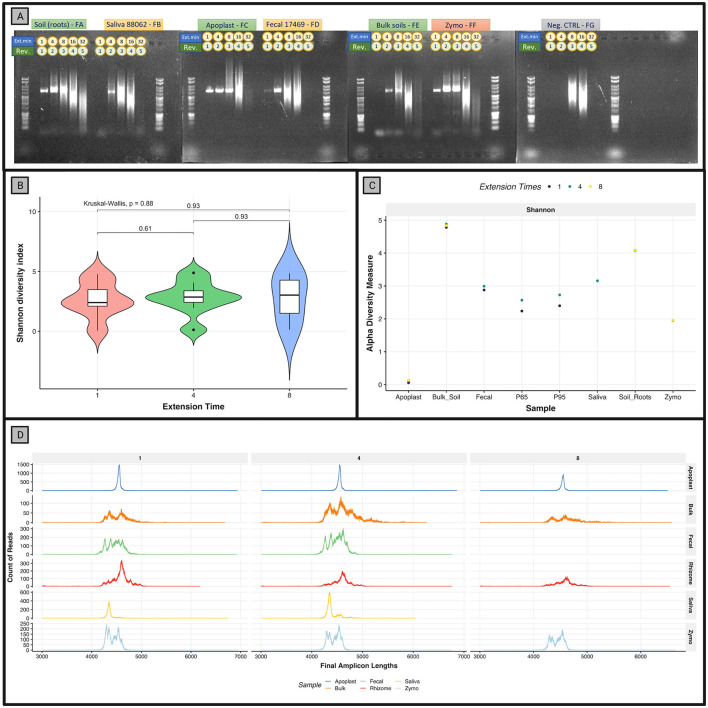
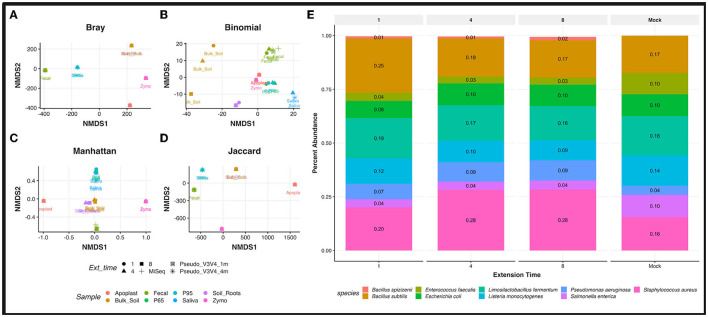
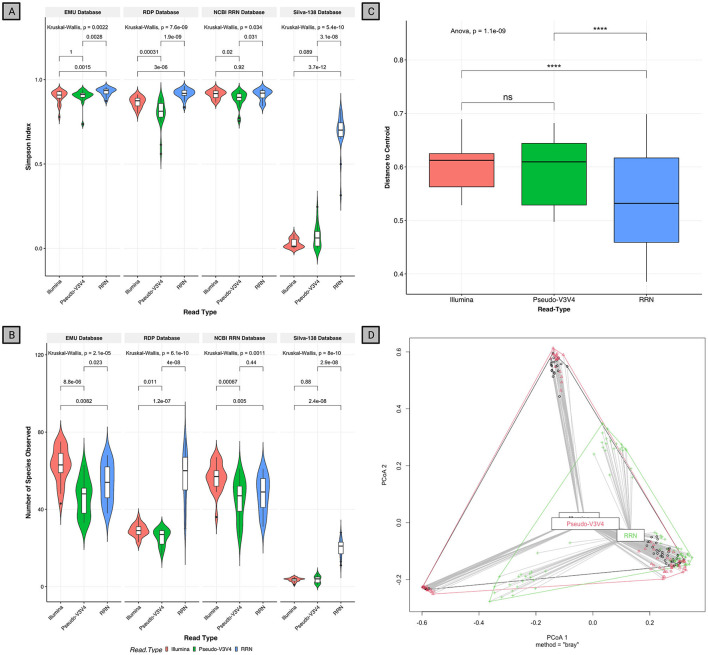
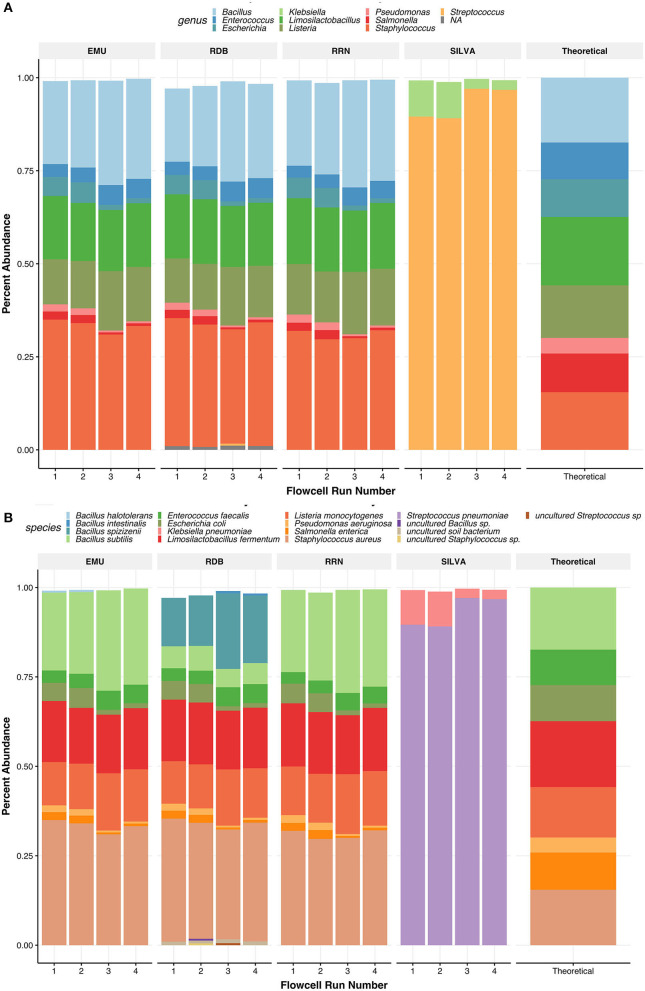
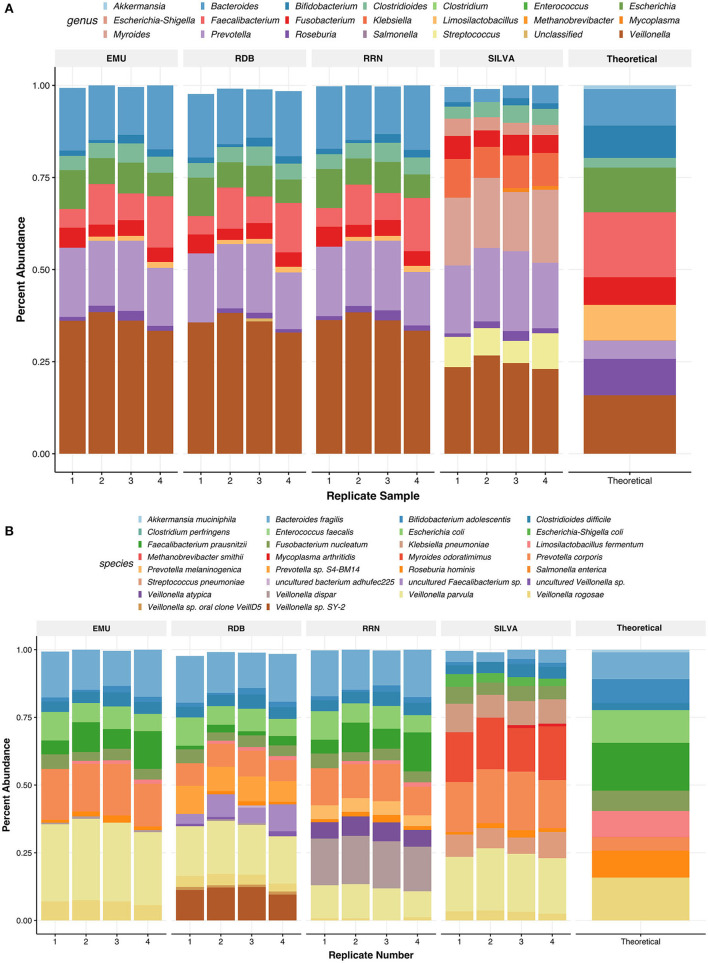
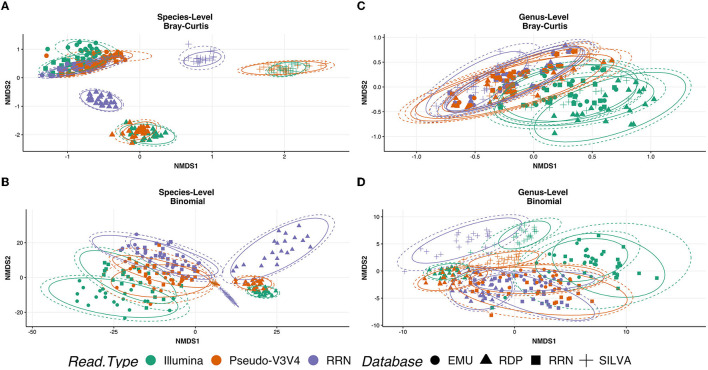
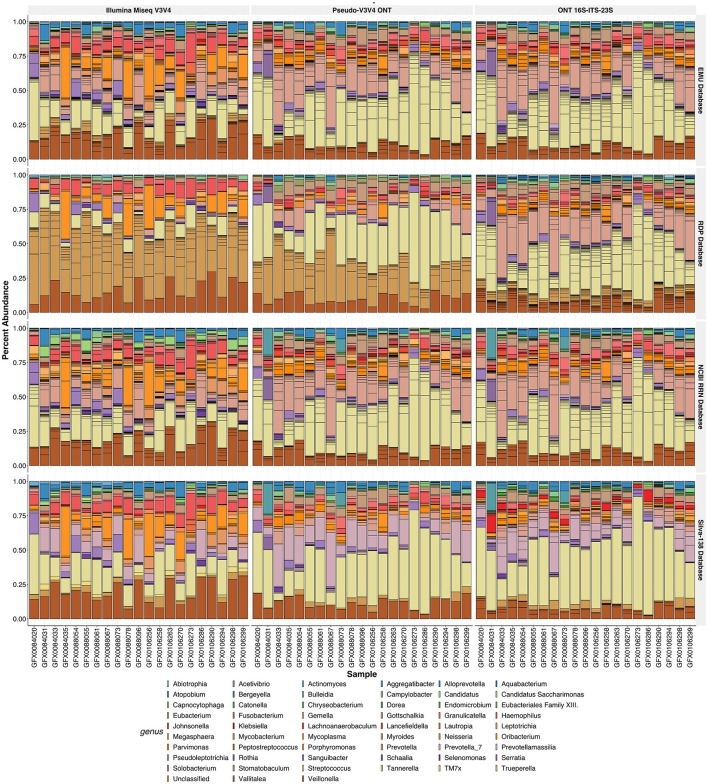
Despite the advent of third-generation sequencing technologies, modern bacterial ecology studies still use Illumina to sequence small (~400 bp) hypervariable regions of the 16S rRNA SSU for phylogenetic classification. By sequencing a larger region of the rRNA gene operons, the limitations and biases of sequencing small portions can be removed, allowing for more accurate classification with deeper taxonomic resolution. With Nanopore sequencing now providing raw simplex reads with quality scores above Q20 using the kit 12 chemistry, the ease, cost, and portability of Nanopore play a leading role in performing differential bacterial abundance analysis. Sequencing the near-entire operon of bacteria and archaea enables the use of the universally conserved operon holding evolutionary polymorphisms for taxonomic resolution. Here, a reproducible and validated pipeline was developed, RRN-operon Enabled Species-level Classification Using EMU (RESCUE), to facilitate the sequencing of bacterial operons and to support import into phyloseq. Benchmarking RESCUE showed that fully processed reads are now parallel or exceed the quality of Sanger, with median quality scores of approximately Q20+, using the R10.4 and Guppy SUP basecalling. The pipeline was validated through two complex mock samples, the use of multiple sample types, with actual Illumina data, and across four databases. RESCUE sequencing is shown to drastically improve classification to the species level for most taxa and resolves erroneous taxa caused by using short reads such as Illumina.
尽管第三代测序技术已经出现,但现代细菌生态学研究仍使用Illumina对16S rRNA小亚基(SSU)的小片段(约400 bp)高变区进行测序,以进行系统发育分类。通过对rRNA基因操纵子的更大区域进行测序,可以消除对小片段测序的局限性和偏差,从而以更深的分类分辨率进行更准确的分类。如今,使用12号试剂盒化学方法,纳米孔测序能够提供质量分数高于Q20的原始单链读数,纳米孔测序的简便性、成本和便携性在进行细菌丰度差异分析中发挥着主导作用。对细菌和古菌的近乎完整的操纵子进行测序,能够利用具有进化多态性的普遍保守操纵子进行分类解析。在此,开发了一种可重复且经过验证的流程,即使用EMU进行rRNA操纵子支持的物种水平分类(RESCUE),以促进细菌操纵子的测序,并支持导入phyloseq。对RESCUE的基准测试表明,使用R10.4和Guppy SUP碱基识别,经过完全处理的读数现在与桑格测序质量相当或超过其质量,中位数质量分数约为Q20+。该流程通过两个复杂的模拟样本、多种样本类型的使用、实际的Illumina数据以及四个数据库进行了验证。结果表明,RESCUE测序能大幅提高大多数分类群到物种水平的分类,并解决因使用Illumina等短读长测序导致的错误分类群问题。