Mitry Madonna M A, Boateng Samuel Y, Greco Francesca, Osborn Helen M I
Reading School of Pharmacy, University of Reading Whiteknights Reading RG6 6AD UK
Dept. of Pharmaceutical Chemistry, Faculty of Pharmacy, Ain Shams University Cairo 11566 Egypt.
RSC Med Chem. 2023 Jul 5;14(8):1537-1548. doi: 10.1039/d3md00137g. eCollection 2023 Aug 16.
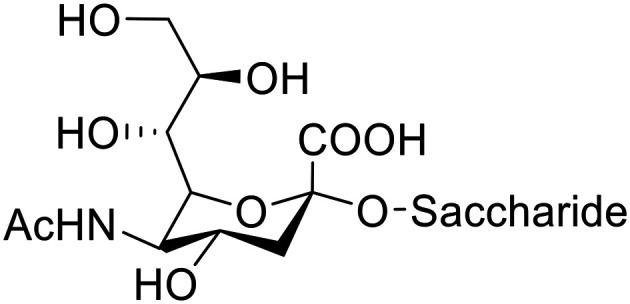
Selective prodrug activation at a tumor site is crucial to maximise the efficiency of chemotherapy approaches and minimise side effects due to off-site activation. In this paper, a new prodrug activation strategy is reported based on the bioorthogonal Staudinger reaction. The feasibility of this prodrug activation strategy was initially demonstrated using 9-azido sialic acid 4 as a trigger and two novel triphenylphosphine-modified N-mustard-PRO 10 and doxorubicin-PRO 12 prodrugs in an HPLC-monitored release study. Then, the azide reporter group was introduced on cancer cells' surfaces through metabolic glycoengineering of sialic acid-rich surface glycans using azide-modified monosaccharides (9-azido sialic acid 4, tetra--acetylated-9-azido sialic acid 5 and tetra--acetyl azidomannosamine). Next, the N-mustard-PRO 10 and doxorubicin-PRO 12 prodrugs were employed with the bioengineered cells, and activation of the prodrugs, which allowed selective release of the cytotoxic moiety at the tumour cell, was assessed. Release of the parent drugs from the prodrugs was shown to be dependent on the level of metabolic labelling, where tetra--acetyl azidomannosamine allowed the highest level of azide reporter generation in tumor cells and led to full recovery of the parent cytotoxic drug's potency. The selectivity of azide expression on breast cancer MCF-7 cells normal fibroblast L929 cells was also probed, with the 9-azido sialic acid and tetra--acetylated-9-azido sialic acid showing ∼17-fold higher azide expression on the former. Taken together, these data demonstrate the feasibility of the Staudinger reaction for selective activation of prodrugs targeted to the MCF-7 breast cancer cells.
在肿瘤部位进行选择性前药激活对于最大化化疗方法的效率以及最小化由于非靶向激活引起的副作用至关重要。本文报道了一种基于生物正交施陶丁格反应的新前药激活策略。该前药激活策略的可行性最初在一项HPLC监测的释放研究中,使用9-叠氮唾液酸4作为触发剂以及两种新型三苯基膦修饰的N-芥子气前药10和阿霉素前药12得以证明。然后,通过使用叠氮修饰的单糖(9-叠氮唾液酸4、四-O-乙酰基-9-叠氮唾液酸5和四-O-乙酰基叠氮甘露糖胺)对富含唾液酸的表面聚糖进行代谢糖工程,将叠氮报告基团引入癌细胞表面。接下来,将N-芥子气前药10和阿霉素前药12用于经过生物工程改造的细胞,并评估前药的激活情况,前药的激活使得细胞毒性部分在肿瘤细胞处选择性释放。结果表明,母体药物从前药中的释放取决于代谢标记水平,其中四-O-乙酰基叠氮甘露糖胺在肿瘤细胞中允许产生最高水平的叠氮报告基团,并导致母体细胞毒性药物的效力完全恢复。还探究了乳腺癌MCF-7细胞与正常成纤维细胞L929细胞上叠氮表达的选择性,9-叠氮唾液酸和四-O-乙酰基-9-叠氮唾液酸在前者上的叠氮表达高出约17倍。综上所述,这些数据证明了施陶丁格反应用于选择性激活靶向MCF-7乳腺癌细胞的前药的可行性。