Stewart Computational Chemistry, 15210 Paddington Circle, Colorado Springs, CO, 80921, USA.
J Mol Model. 2023 Aug 22;29(9):284. doi: 10.1007/s00894-023-05695-1.
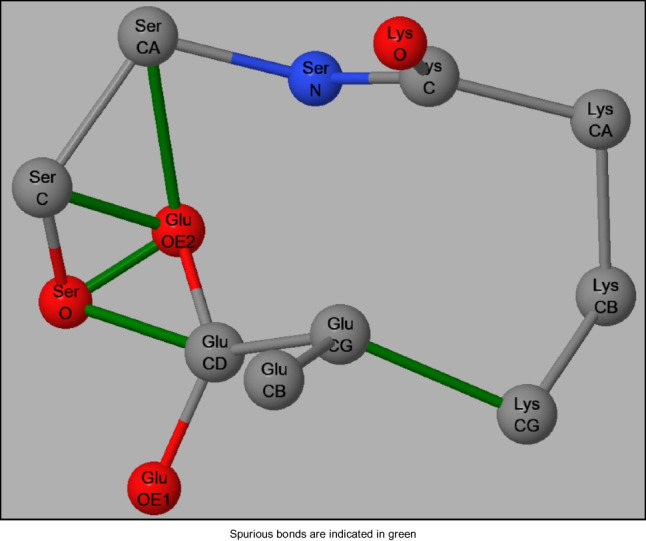
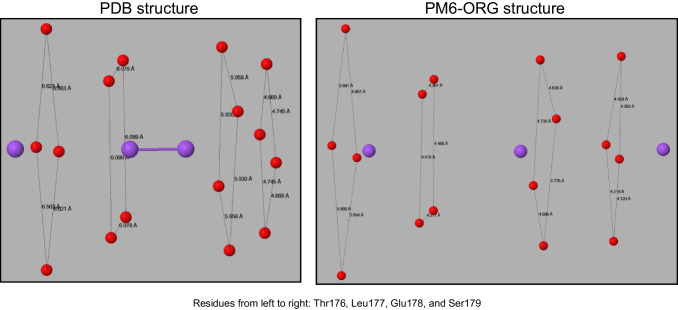
In recent years, semiempirical methods such as PM6, PM6-D3H4, and PM7 have been increasingly used for modeling proteins, in particular enzymes. These methods were designed for more general use, and consequently were not optimized for studying proteins. Because of this, various specific errors have been found that could potentially cast doubt on the validity of these methods for modeling phenomena of biochemical interest such as enzyme catalytic mechanisms and protein-ligand interactions. To correct these and other errors, a new method specifically designed for use in organic and biochemical modeling has been developed.
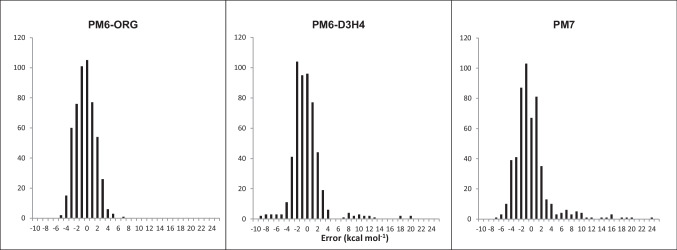
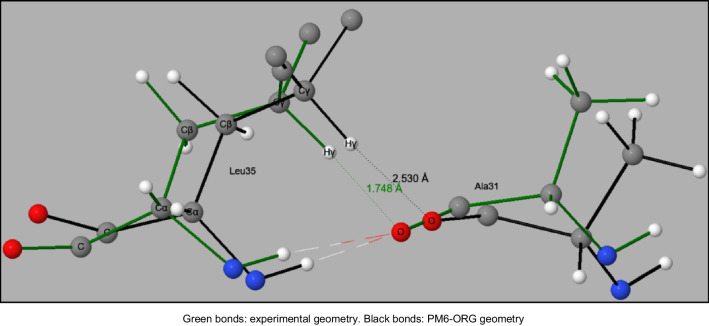
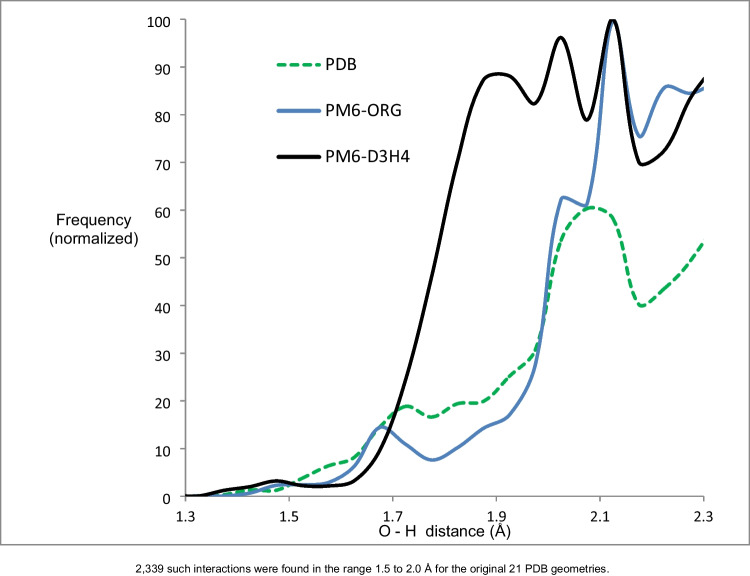
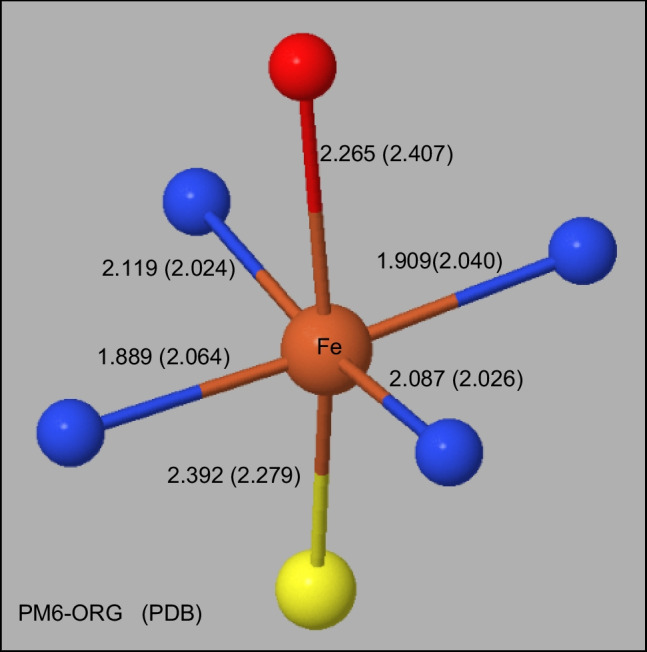
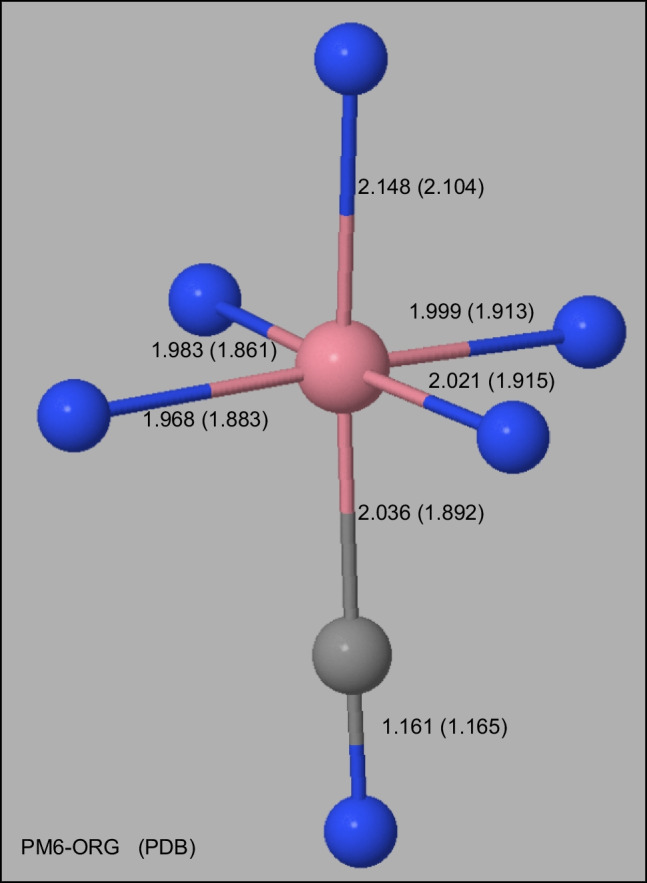
Two alterations were made to the procedures used in developing the earlier PMx methods. A minor change was made to the theoretical framework, which affected only the non-quantum theory interatomic interaction function, while the major change involved changing the training set for optimizing parameters, moving the focus to systems of biochemical significance. This involved both the selection of reference data and the weighting factors, i.e., the relative importance that the various data were given. As a result of this change of focus, the accuracy in prediction of heats of formation, hydrogen bonding, and geometric quantities relating to non-covalent interactions in proteins was improved significantly.
近年来,PM6、PM6-D3H4 和 PM7 等半经验方法已越来越多地用于蛋白质建模,尤其是酶的建模。这些方法的设计初衷是更通用的用途,因此并未针对蛋白质研究进行优化。正因为如此,人们发现了各种特定的错误,这些错误可能会对这些方法用于模拟生化相关现象(如酶催化机制和蛋白配体相互作用)的有效性产生怀疑。为了纠正这些和其他错误,专门为有机和生化建模设计了一种新方法。
对早期 PMx 方法中使用的程序进行了两项修改。理论框架进行了微小的改变,仅影响非量子理论的原子间相互作用函数,而主要的改变涉及优化参数的训练集,将重点转移到具有生化意义的系统。这涉及参考数据和权重因子的选择,即对各种数据赋予的相对重要性。由于重点的这种改变,蛋白质中非共价相互作用的生成热、氢键和几何量的预测准确性得到了显著提高。