Qiu Yuchi, Wei Guo-Wei
Department of Mathematics, Michigan State University, East Lansing, MI 48824, USA.
Department of Biochemistry and Molecular Biology, Michigan State University, MI, 48824, USA.
Nat Comput Sci. 2023 Feb;3(2):149-163. doi: 10.1038/s43588-022-00394-y. Epub 2023 Feb 20.
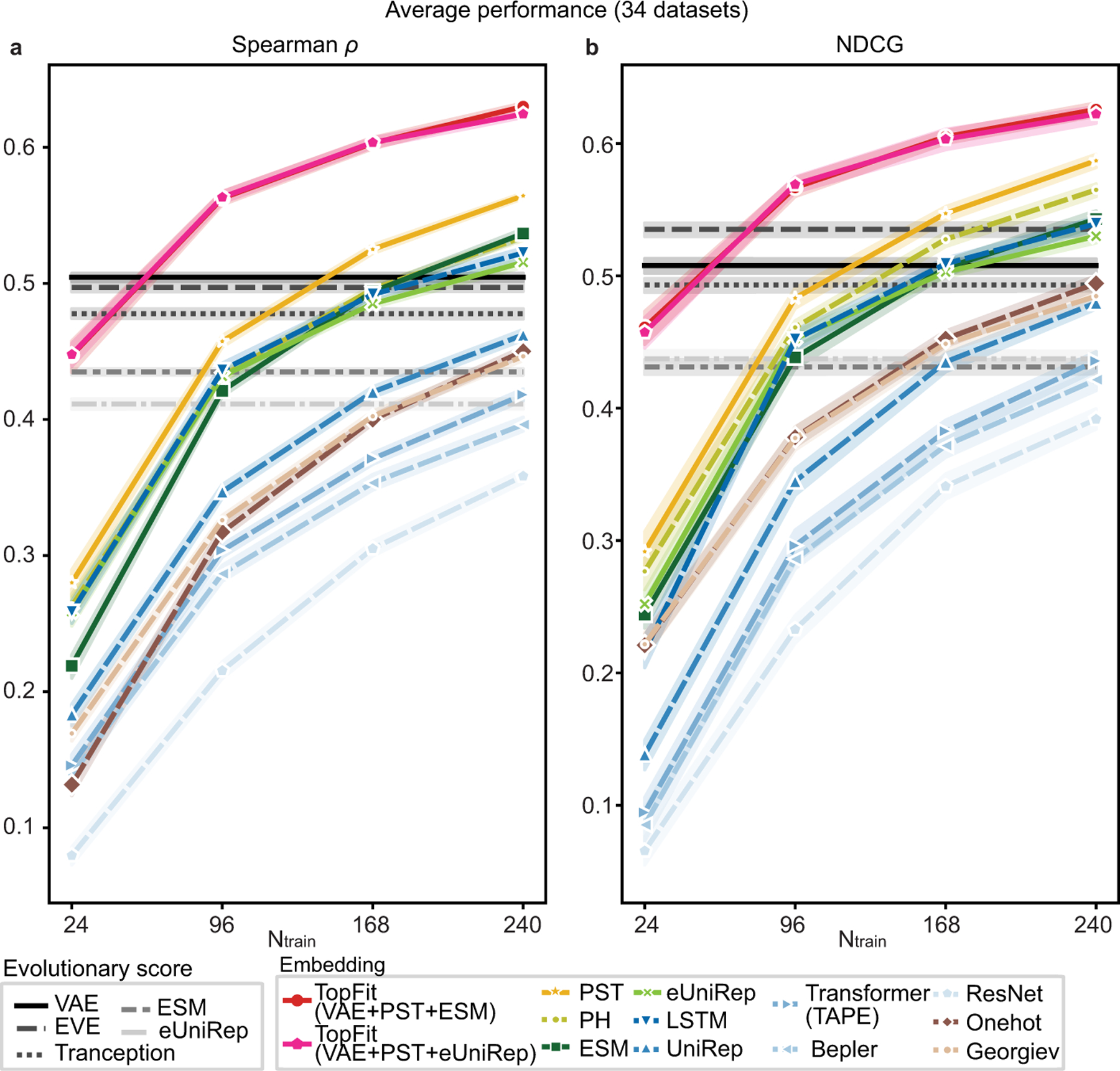
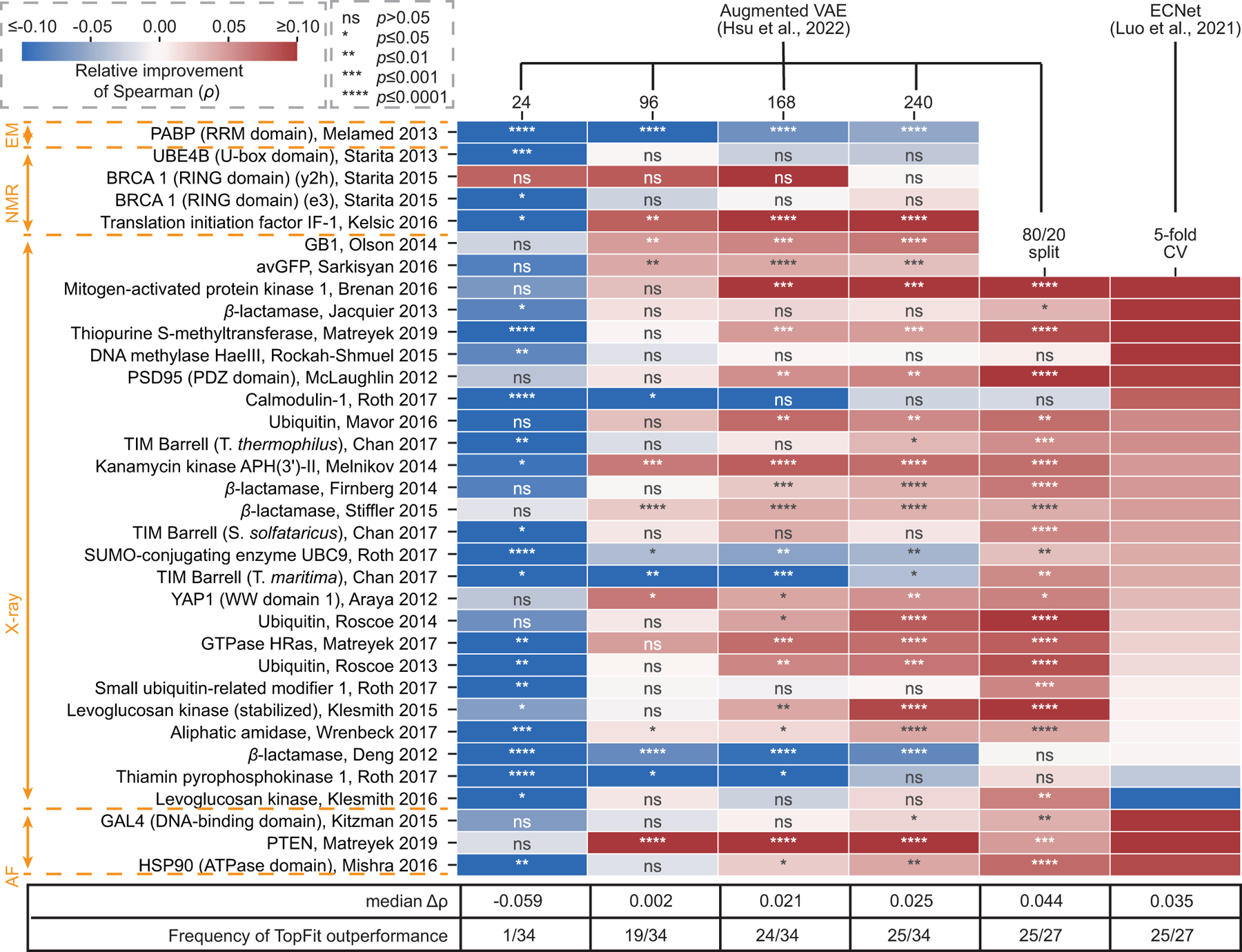
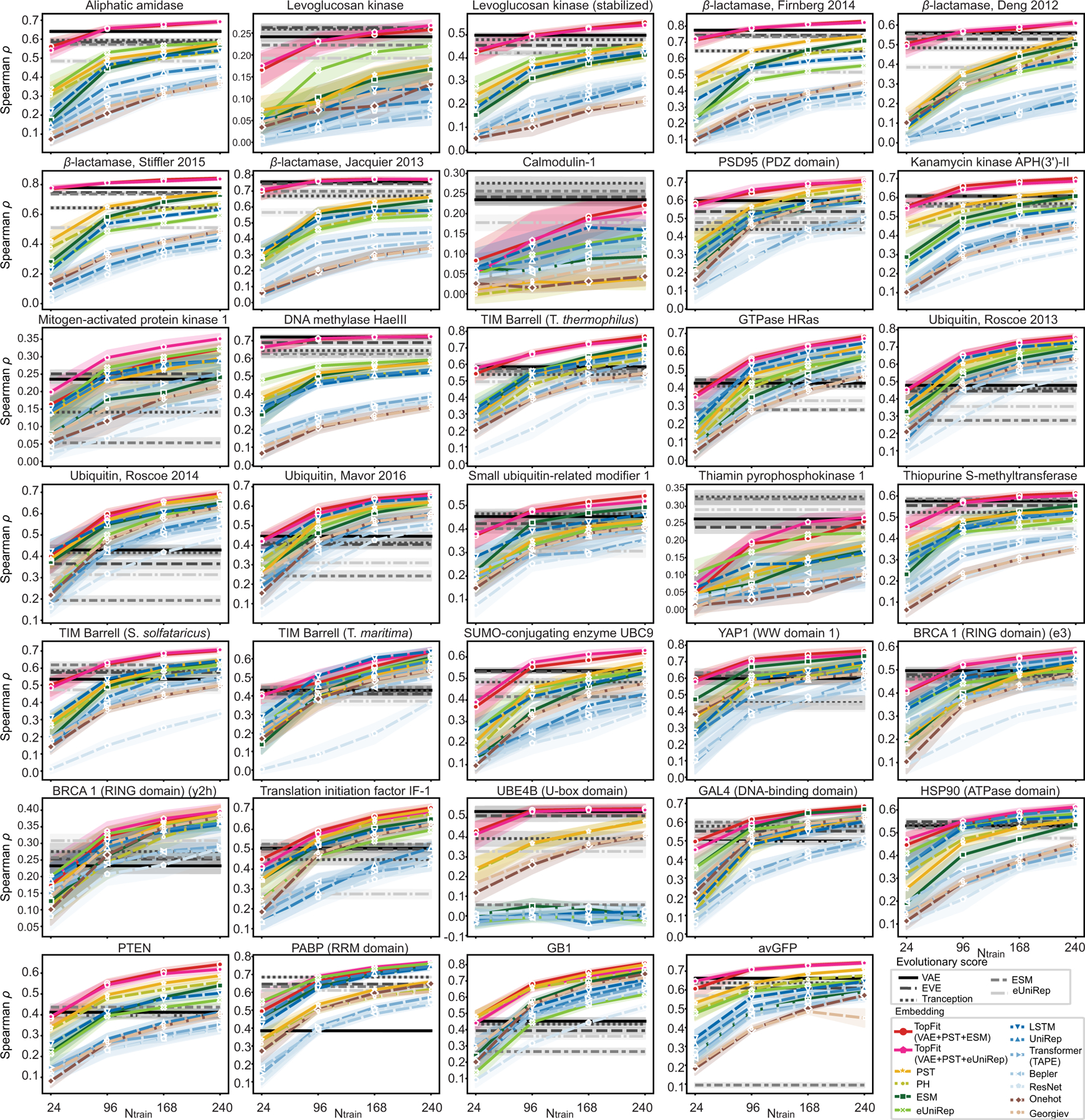
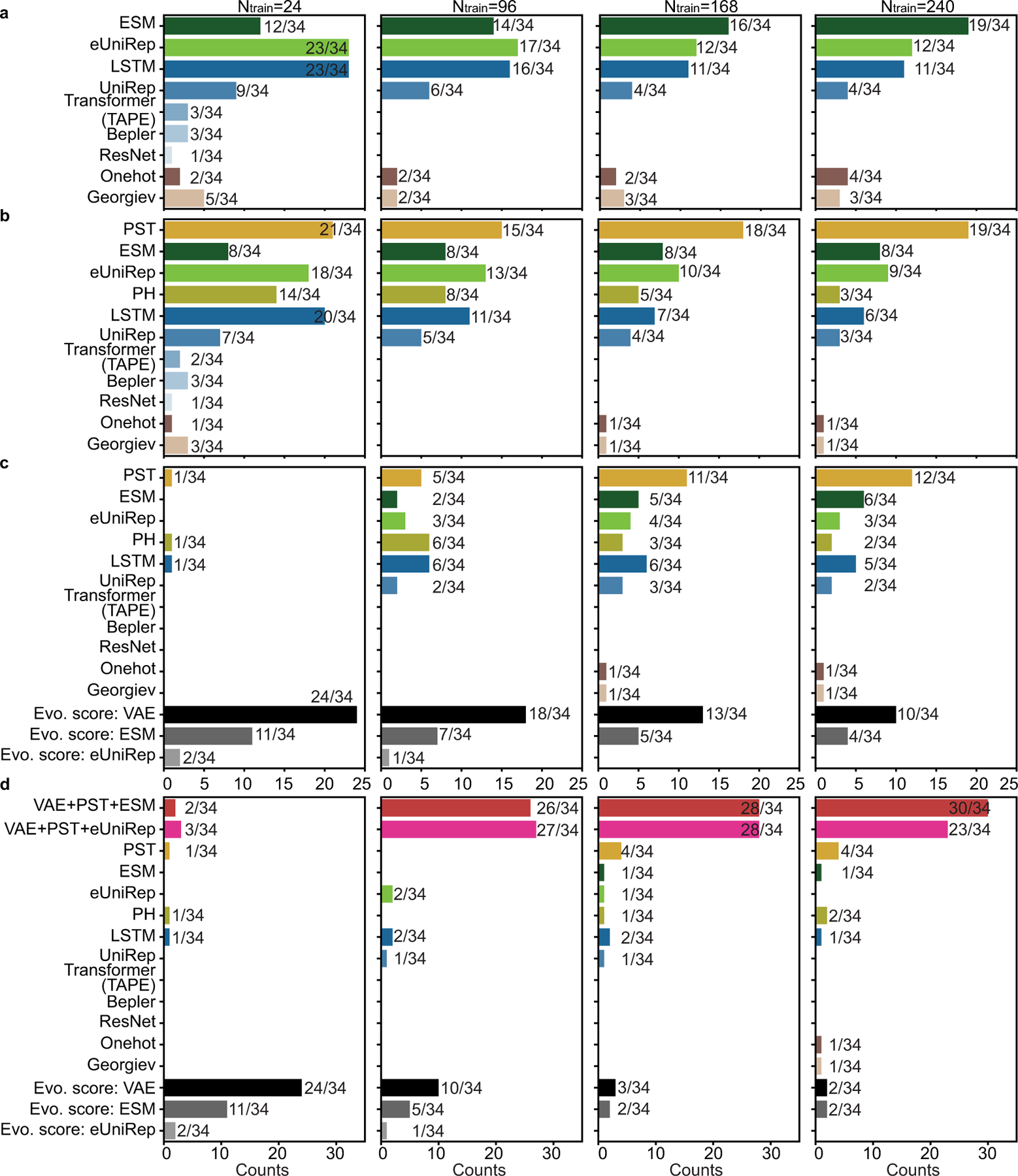
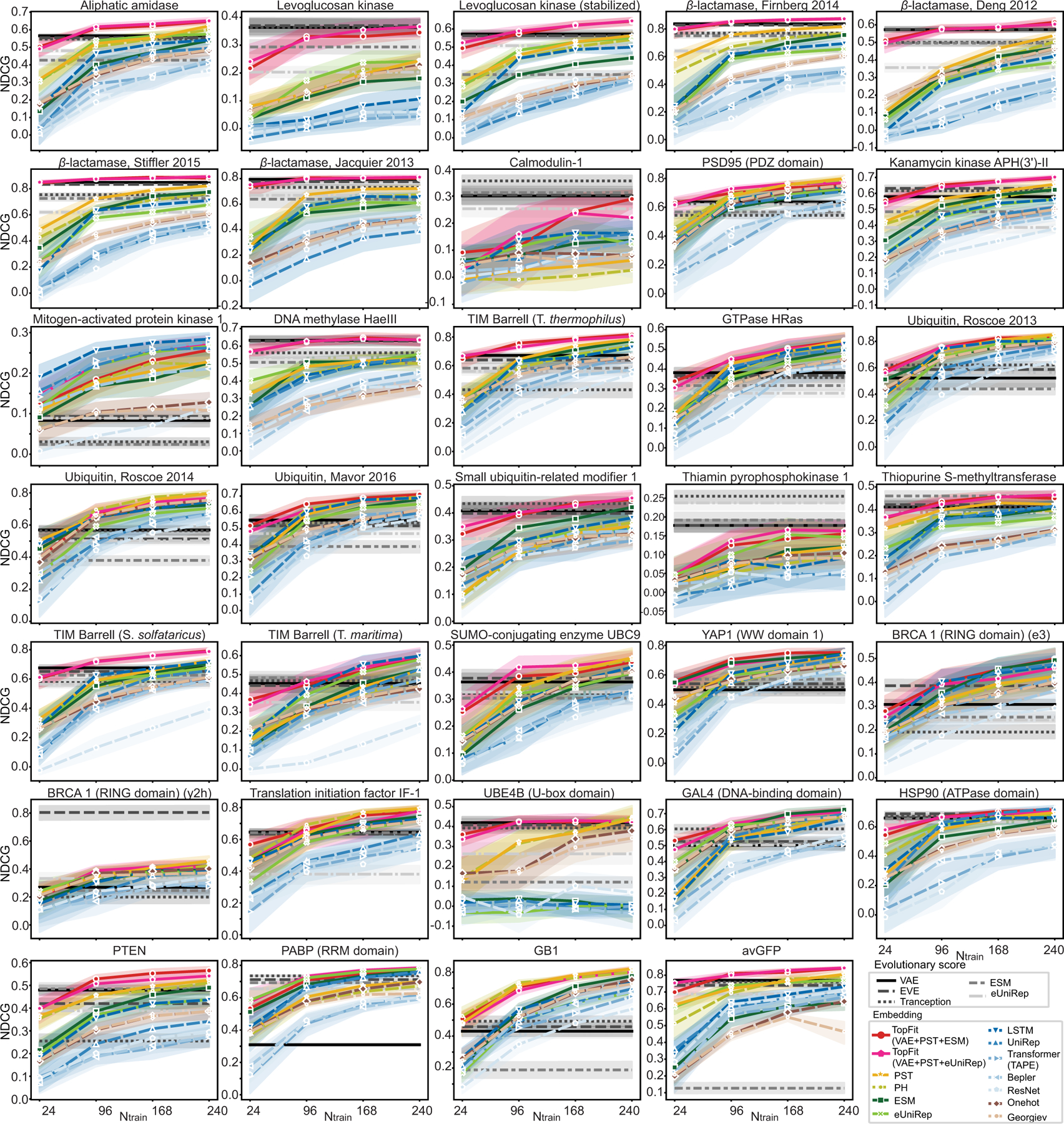
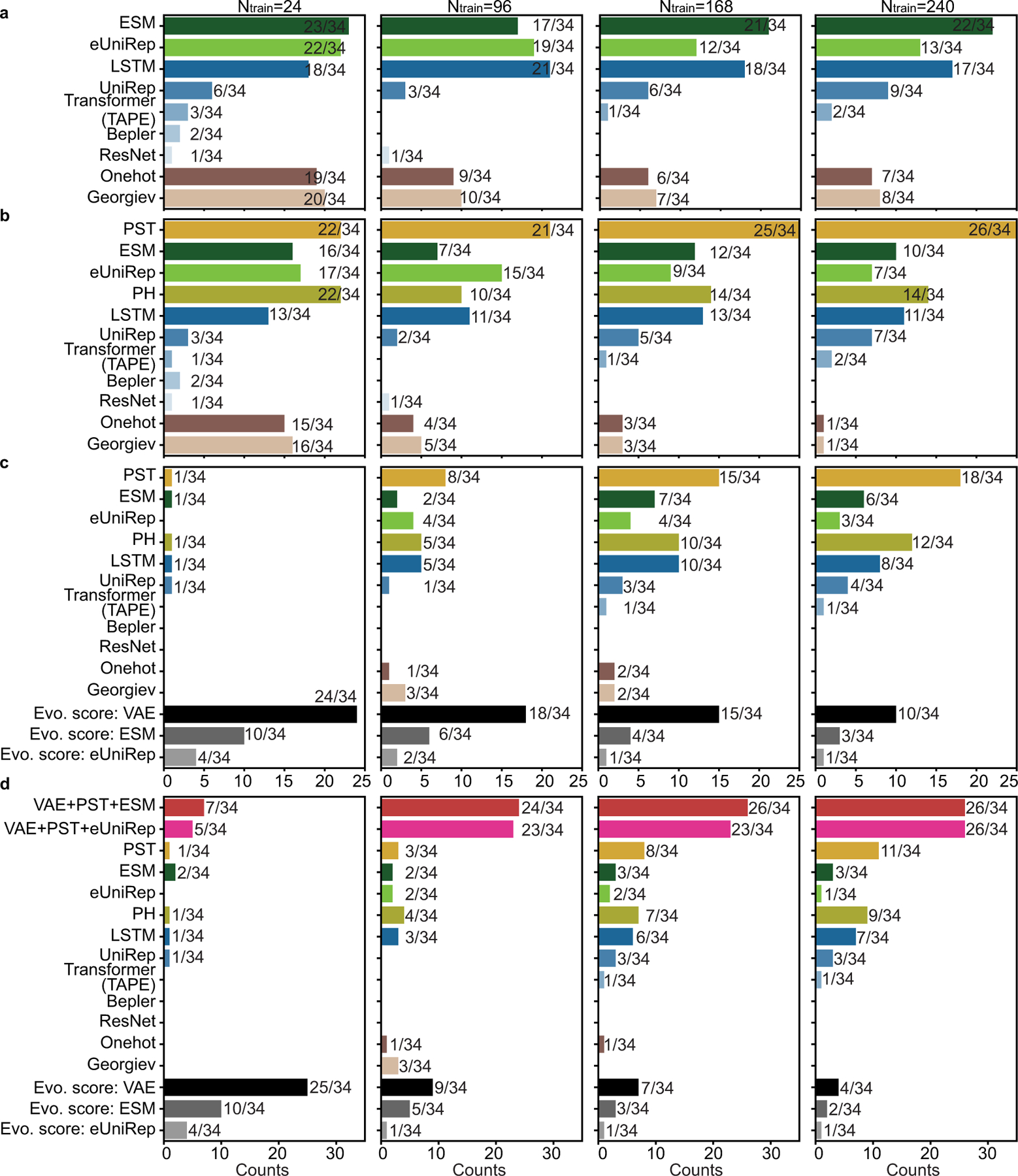
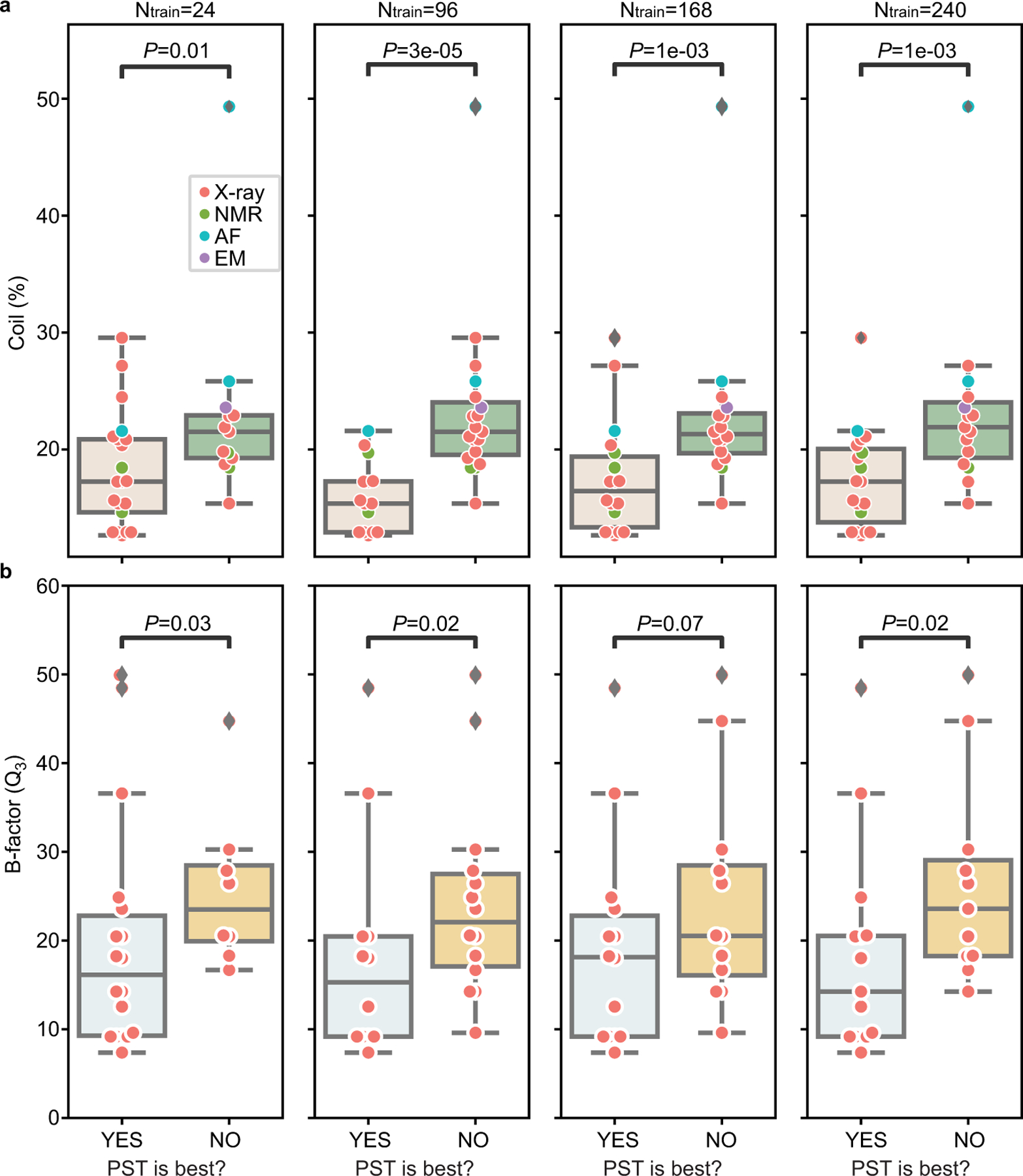
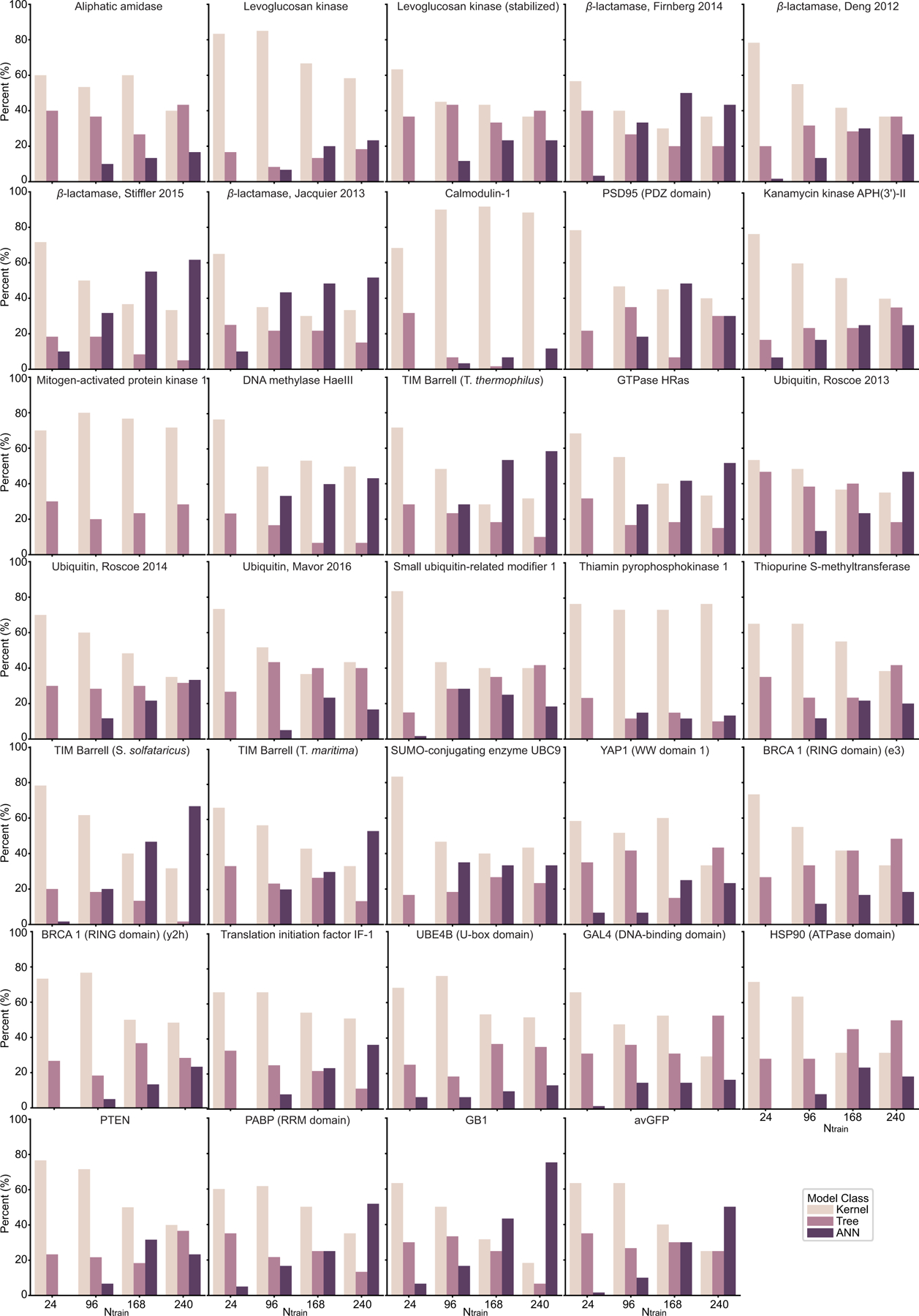
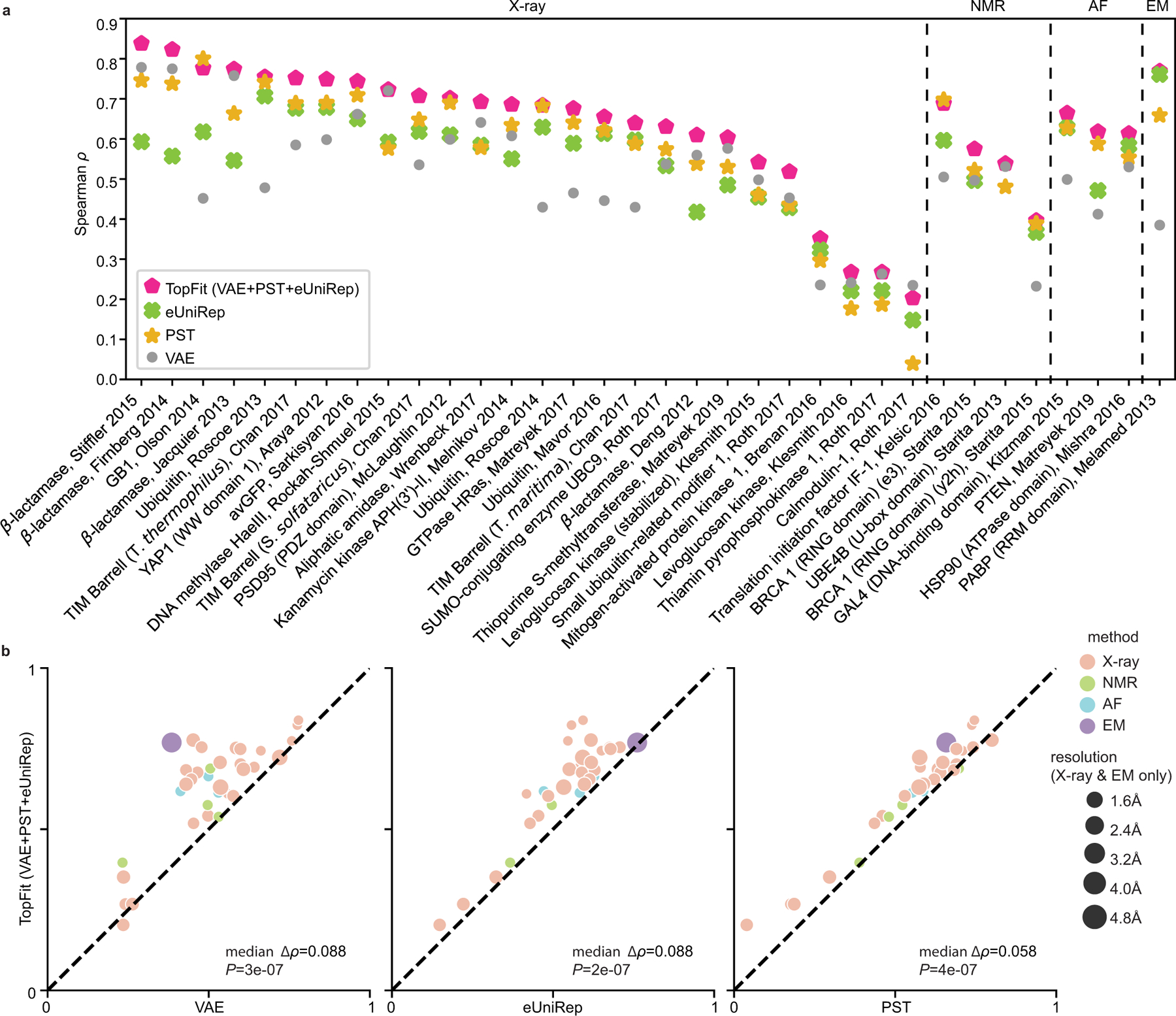
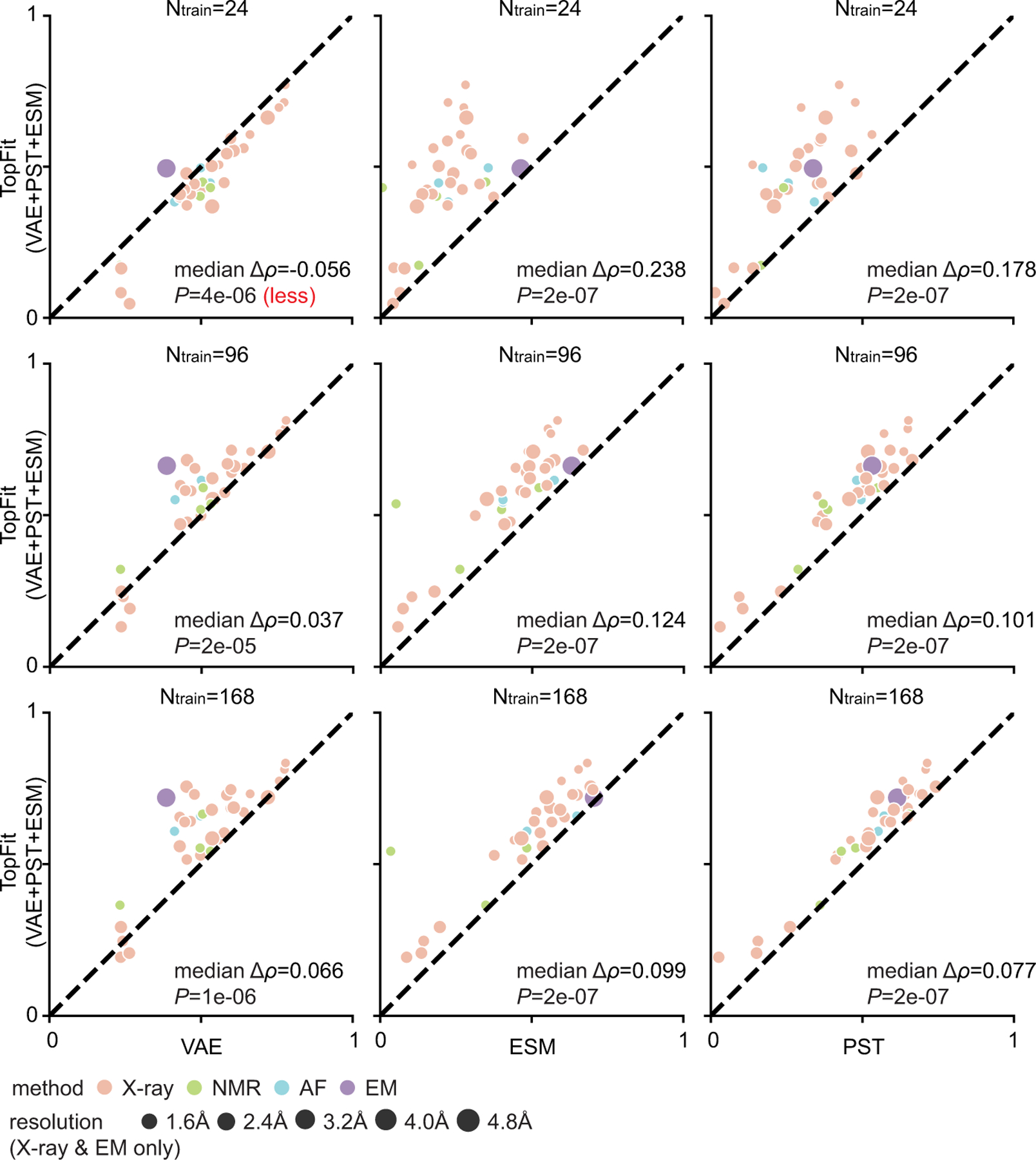
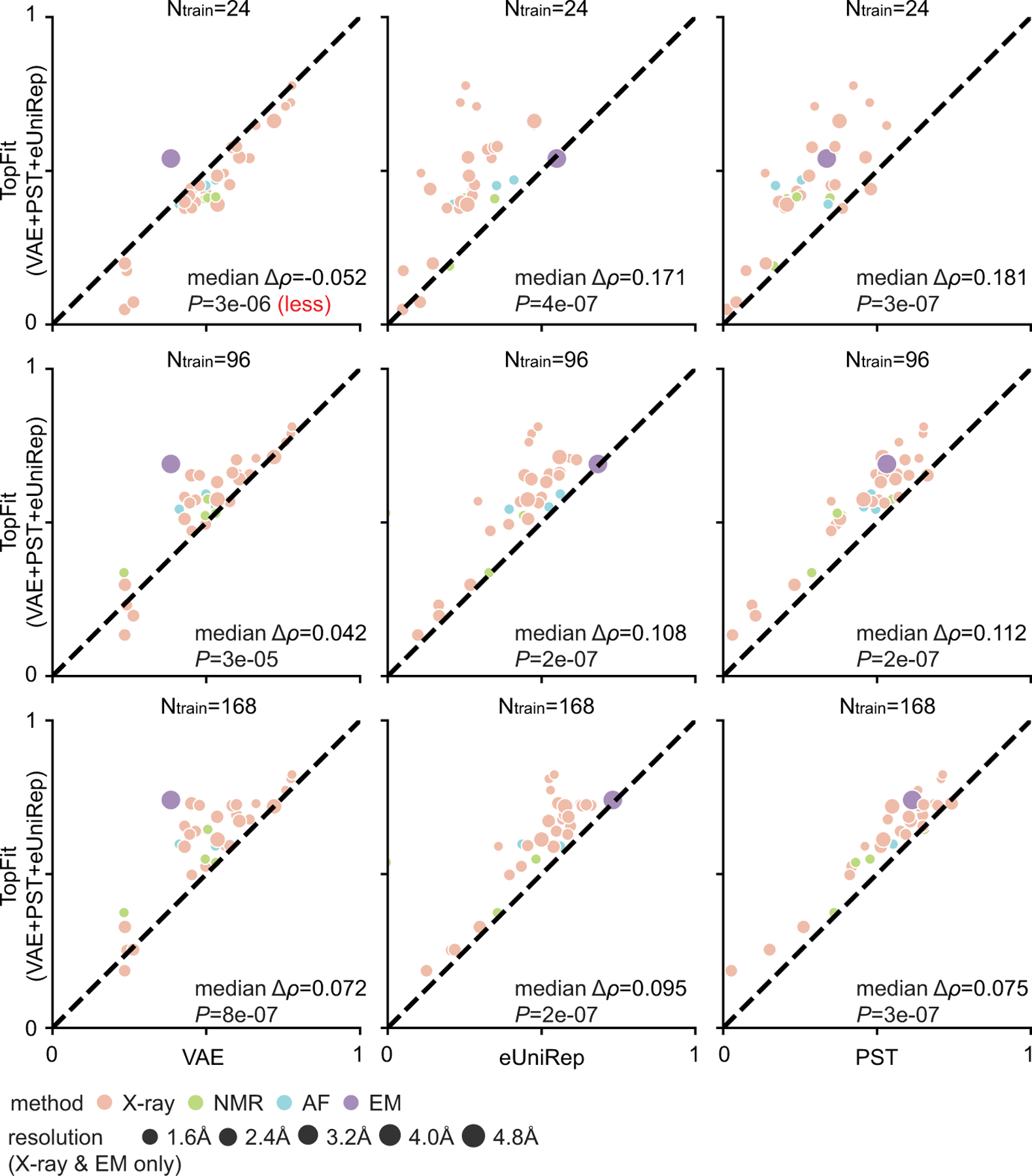
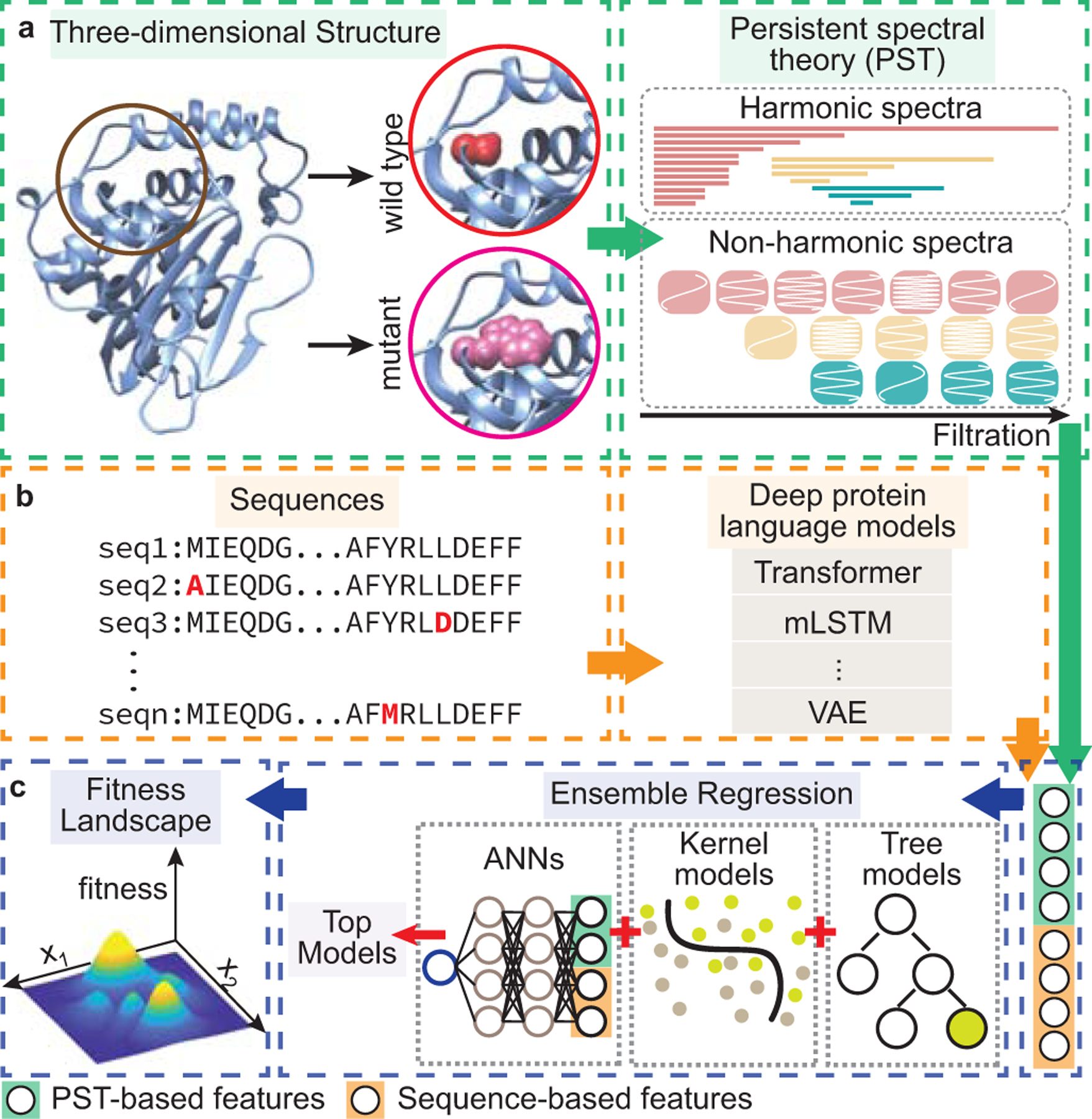
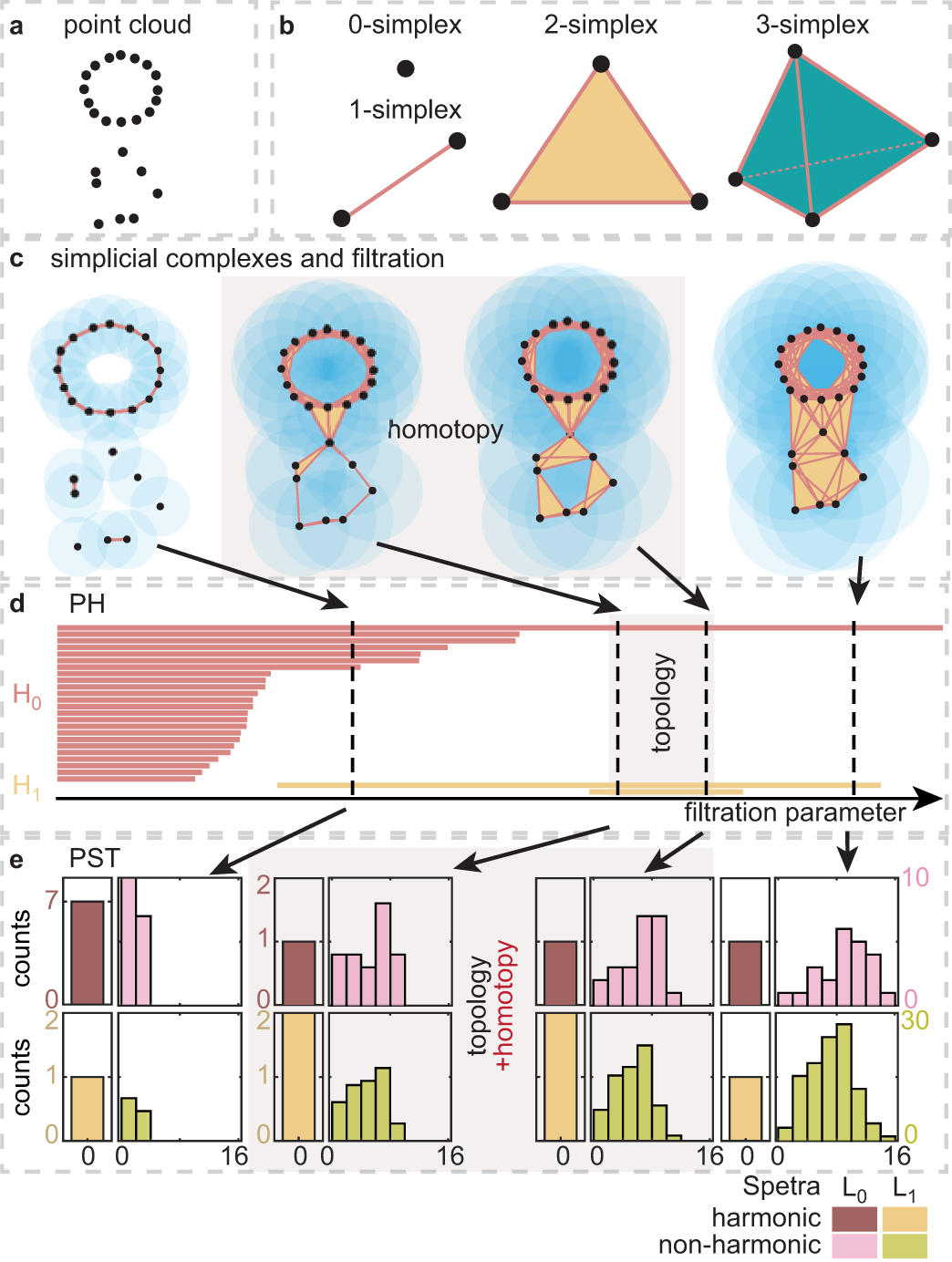
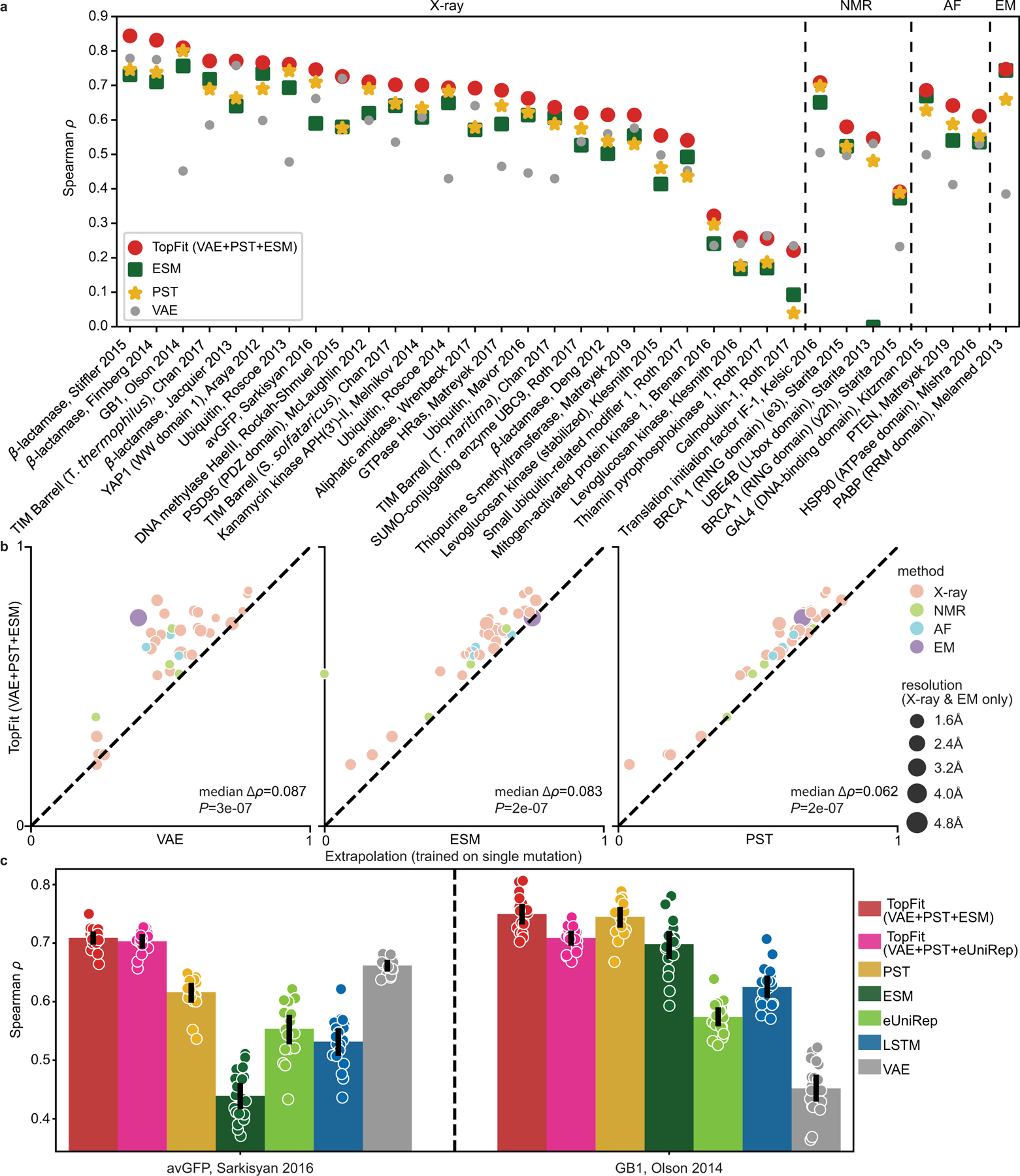
While protein engineering, which iteratively optimizes protein fitness by screening the gigantic mutational space, is constrained by experimental capacity, various machine learning models have substantially expedited protein engineering. Three-dimensional protein structures promise further advantages, but their intricate geometric complexity hinders their applications in deep mutational screening. Persistent homology, an established algebraic topology tool for protein structural complexity reduction, fails to capture the homotopic shape evolution during the filtration of a given data. This work introduces a opology-ffered rotein ness (TopFit) framework to complement protein sequence and structure embeddings. Equipped with an ensemble regression strategy, TopFit integrates the persistent spectral theory, a new topological Laplacian, and two auxiliary sequence embeddings to capture mutation-induced topological invariant, shape evolution, and sequence disparity in the protein fitness landscape. The performance of TopFit is assessed by 34 benchmark datasets with 128,634 variants, involving a vast variety of protein structure acquisition modalities and training set size variations.
虽然通过筛选巨大的突变空间来迭代优化蛋白质适应性的蛋白质工程受到实验能力的限制,但各种机器学习模型已大大加快了蛋白质工程的进程。三维蛋白质结构具有进一步的优势,但其复杂的几何复杂性阻碍了它们在深度突变筛选中的应用。持久同调作为一种用于降低蛋白质结构复杂性的既定代数拓扑工具,在给定数据的过滤过程中无法捕捉到同伦形状的演变。这项工作引入了一种拓扑提供的蛋白质适应性(TopFit)框架,以补充蛋白质序列和结构嵌入。配备了集成回归策略,TopFit整合了持久谱理论、一种新的拓扑拉普拉斯算子和两个辅助序列嵌入,以捕捉蛋白质适应性景观中突变诱导的拓扑不变性、形状演变和序列差异。通过34个包含128,634个变体的基准数据集评估了TopFit的性能,这些数据集涉及各种各样的蛋白质结构获取方式和训练集大小变化。