Wee JunJie, Wei Guo-Wei
Department of Mathematics, Michigan State University, East Lansing, MI 48824, USA.
Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI 48824, USA.
ArXiv. 2024 Nov 19:arXiv:2411.12370v1.
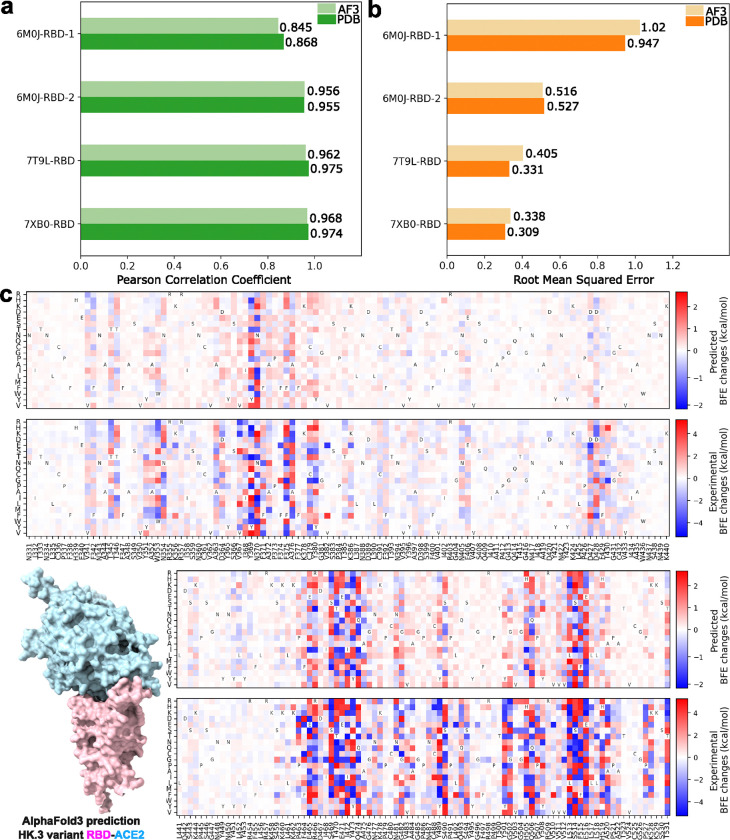
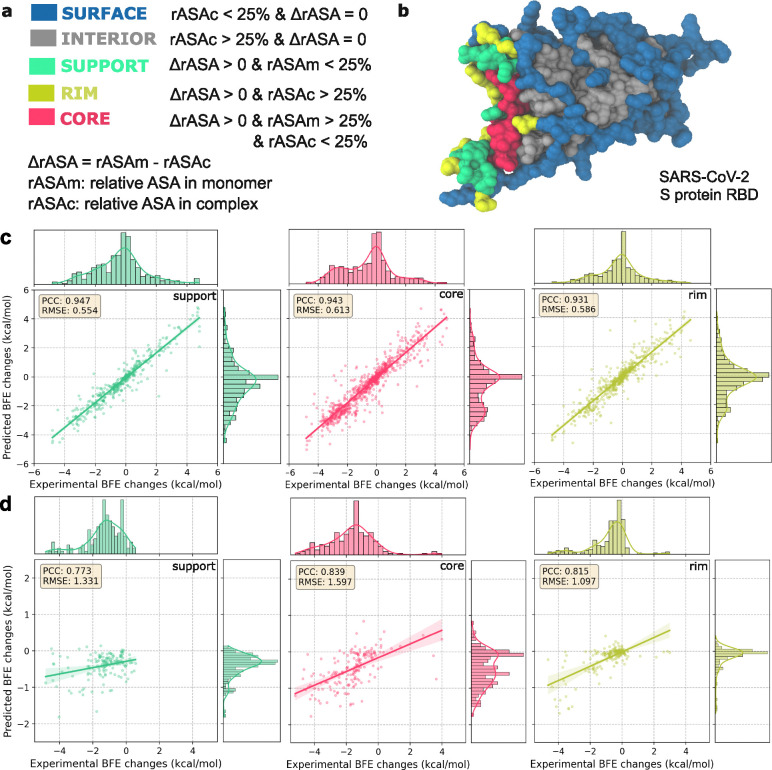
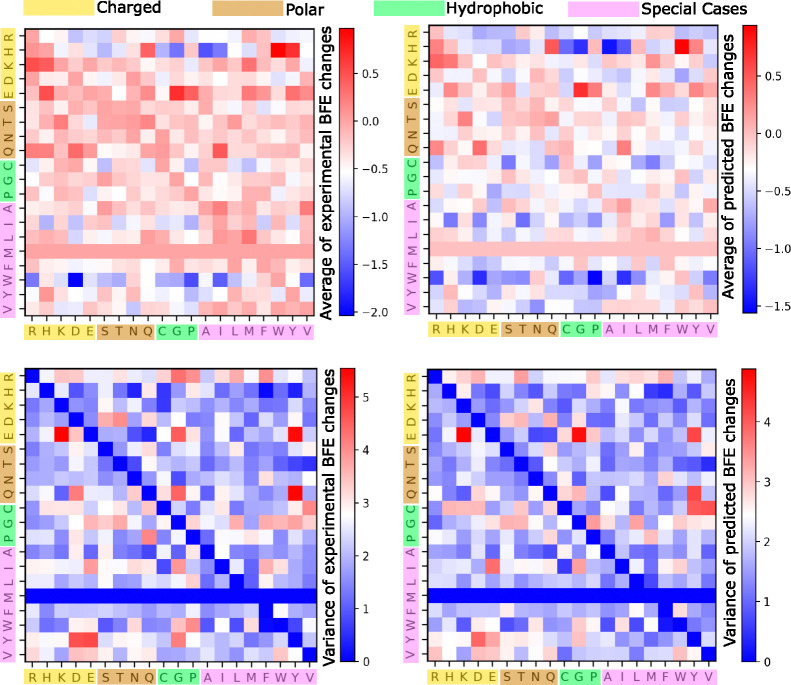
The fast evolution of SARS-CoV-2 and other infectious viruses poses a grand challenge to the rapid response in terms of viral tracking, diagnostics, and design and manufacture of monoclonal antibodies (mAbs) and vaccines, which are both time-consuming and costly. This underscores the need for efficient computational approaches. Recent advancements, like topological deep learning (TDL), have introduced powerful tools for forecasting emerging dominant variants, yet they require deep mutational scanning (DMS) of viral surface proteins and associated three-dimensional (3D) protein-protein interaction (PPI) complex structures. We propose an AlphaFold 3 (AF3)-assisted multi-task topological Laplacian (MT-TopLap) strategy to address this need. MT-TopLap combines deep learning with topological data analysis (TDA) models, such as persistent Laplacians (PL) to extract detailed topological and geometric characteristics of PPIs, thereby enhancing the prediction of DMS and binding free energy (BFE) changes upon virus mutations. Validation with four experimental DMS datasets of SARS-CoV-2 spike receptor-binding domain (RBD) and the human angiotensin-converting enzyme-2 (ACE2) complexes indicates that our AF3 assisted MT-TopLap strategy maintains robust performance, with only an average 1.1% decrease in Pearson correlation coefficients (PCC) and an average 9.3% increase in root mean square errors (RMSE), compared with the use of experimental structures. Additionally, AF3-assisted MT-TopLap achieved a PCC of 0.81 when tested with a SARS-CoV-2 HK.3 variant DMS dataset, confirming its capability to accurately predict BFE changes and adapt to new experimental data, thereby showcasing its potential for rapid and effective response to fast viral evolution.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)和其他传染性病毒的快速进化,在病毒追踪、诊断以及单克隆抗体(mAb)和疫苗的设计与制造方面对快速响应构成了巨大挑战,这些过程既耗时又昂贵。这凸显了高效计算方法的必要性。最近的进展,如拓扑深度学习(TDL),引入了强大的工具来预测新兴的优势变体,但它们需要对病毒表面蛋白和相关的三维(3D)蛋白质-蛋白质相互作用(PPI)复合物结构进行深度突变扫描(DMS)。我们提出了一种基于阿尔法折叠3(AF3)辅助的多任务拓扑拉普拉斯(MT-TopLap)策略来满足这一需求。MT-TopLap将深度学习与拓扑数据分析(TDA)模型相结合,如持久拉普拉斯(PL),以提取PPI的详细拓扑和几何特征,从而增强对DMS以及病毒突变时结合自由能(BFE)变化的预测。使用四个SARS-CoV-2刺突受体结合域(RBD)和人血管紧张素转换酶2(ACE2)复合物的实验性DMS数据集进行验证表明,与使用实验结构相比,我们的AF3辅助MT-TopLap策略保持了强大的性能,皮尔逊相关系数(PCC)平均仅下降1.1%,均方根误差(RMSE)平均增加9.3%。此外,AF3辅助的MT-TopLap在使用SARS-CoV-2 HK.3变体DMS数据集进行测试时达到了0.81的PCC,证实了其准确预测BFE变化并适应新实验数据的能力,从而展示了其对快速病毒进化进行快速有效响应的潜力。