INSERM, Myology Research Center-UMRS974, Hôpital Universitaire de la Pitié-Salpêtrière, Institut de Myologie, Sorbonne Université, 105 Boulevard de l'Hôpital, 75013, Paris, France.
Service de Neuromyologie, Centre de Référence Neuromusculaire, APHP, Paris, France.
Sci Rep. 2023 Aug 28;13(1):14054. doi: 10.1038/s41598-023-41008-5.
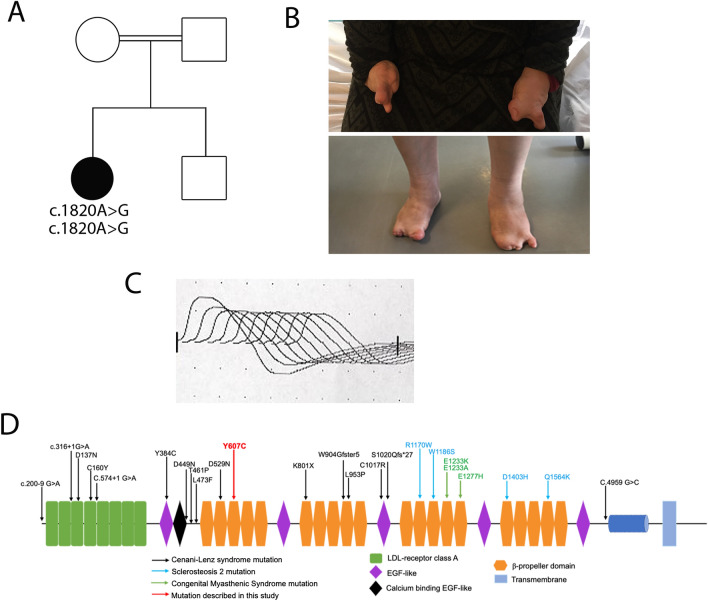
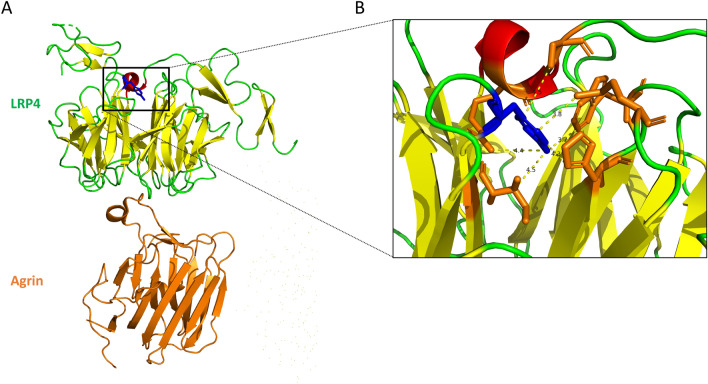
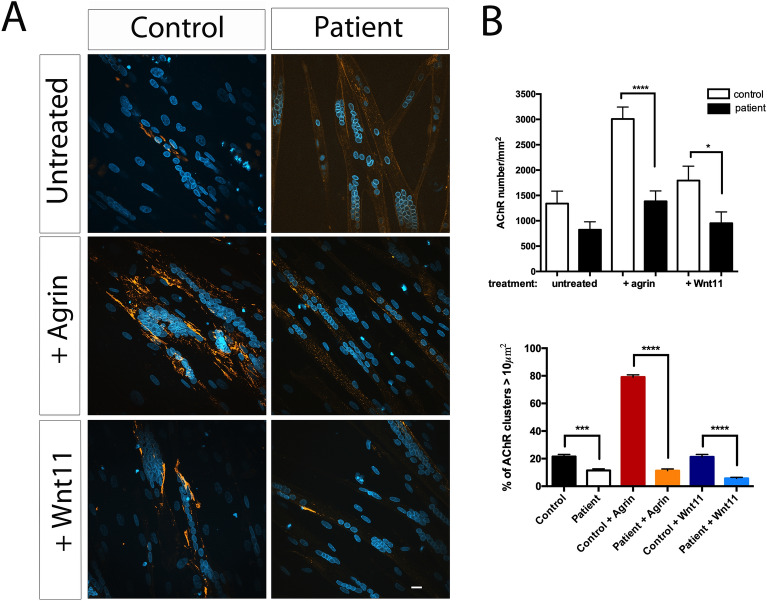
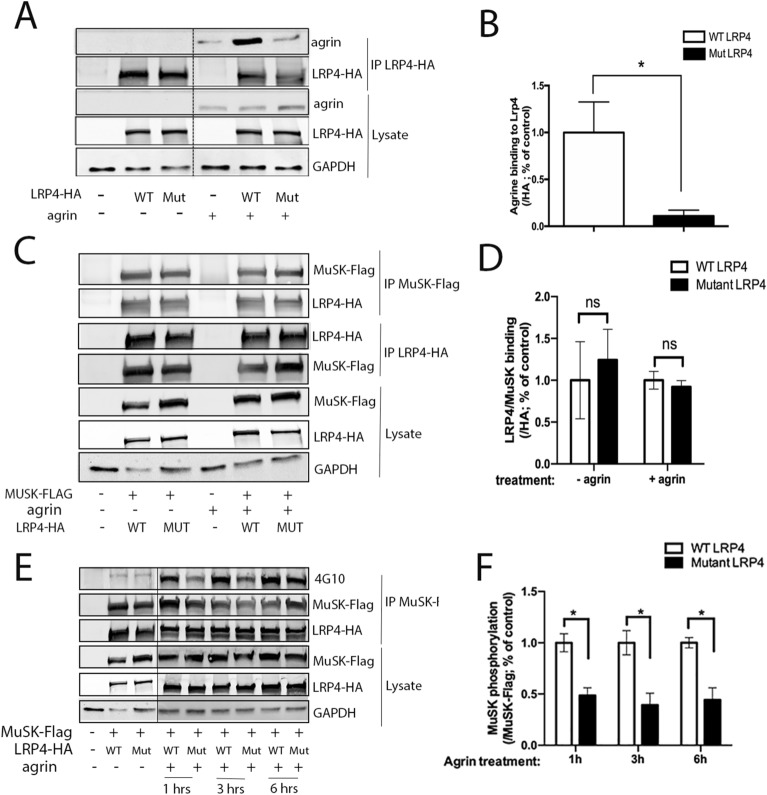
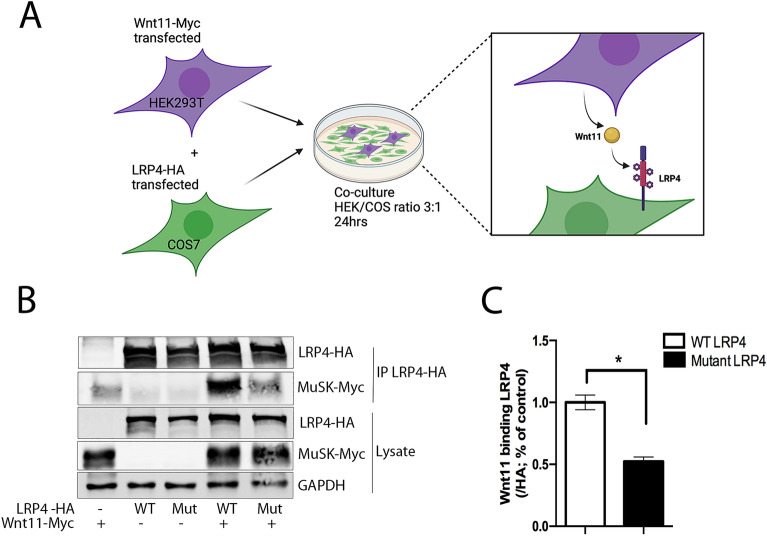
Congenital myasthenic syndromes (CMS) are a clinically and genetically heterogeneous group of rare diseases due to mutations in neuromuscular junction (NMJ) protein-coding genes. Until now, many mutations encoding postsynaptic proteins as Agrin, MuSK and LRP4 have been identified as responsible for increasingly complex CMS phenotypes. The majority of mutations identified in LRP4 gene causes bone diseases including CLS and sclerosteosis-2 and rare cases of CMS with mutations in LRP4 gene has been described so far. In the French cohort of CMS patients, we identified a novel LRP4 homozygous missense mutation (c.1820A > G; p.Thy607Cys) within the β1 propeller domain in a patient presenting CMS symptoms, including muscle weakness, fluctuating fatigability and a decrement in compound muscle action potential in spinal accessory nerves, associated with congenital agenesis of the hands and feet and renal malformation. Mechanistic expression studies show a significant decrease of AChR aggregation in cultured patient myotubes, as well as altered in vitro binding of agrin and Wnt11 ligands to the mutated β1 propeller domain of LRP4 explaining the dual phenotype characterized clinically and electoneuromyographically in the patient. These results expand the LRP4 mutations spectrum associated with a previously undescribed clinical association involving impaired neuromuscular transmission and limb deformities and highlighting the critical role of a yet poorly described domain of LRP4 at the NMJ. This study raises the question of the frequency of this rare neuromuscular form and the future diagnosis and management of these cases.
先天性肌无力综合征(CMS)是一组由于神经肌肉接头(NMJ)蛋白编码基因突变引起的具有临床和遗传异质性的罕见疾病。到目前为止,许多编码突触后蛋白的突变,如 Agrin、MuSK 和 LRP4,已被确定为导致越来越复杂的 CMS 表型的原因。LRP4 基因中大多数已鉴定的突变导致骨骼疾病,包括 CLS 和骨硬化症-2,并且到目前为止,已经描述了 LRP4 基因突变导致罕见 CMS 的病例。在法国 CMS 患者队列中,我们在一名表现出 CMS 症状的患者中发现了一个新的 LRP4 纯合错义突变(c.1820A>G;p.Thy607Cys),位于β1 推进器结构域内,该患者存在肌肉无力、波动性疲劳和副神经复合肌肉动作电位衰减,伴有手和脚先天性发育不全和肾脏畸形。机制表达研究表明,在培养的患者肌管中 AChR 聚集显著减少,以及 Agrin 和 Wnt11 配体与 LRP4 的突变β1 推进器结构域的体外结合改变,解释了该患者临床和电神经肌图上的双重表型。这些结果扩展了与以前未描述的临床关联相关的 LRP4 突变谱,该关联涉及神经肌肉传递受损和肢体畸形,并突出了 LRP4 中尚未描述的结构域在 NMJ 中的关键作用。这项研究提出了这种罕见神经肌肉形式的频率问题以及这些病例的未来诊断和管理问题。