Desai Heta, Ofori Samuel, Boatner Lisa, Yu Fengchao, Villanueva Miranda, Ung Nicholas, Nesvizhskii Alexey I, Backus Keriann
Biological Chemistry Department, David Geffen School of Medicine, UCLA, Los Angeles, CA, 90095, USA.
Molecular Biology Institute, UCLA, Los Angeles, CA, 90095, USA.
bioRxiv. 2023 Aug 14:2023.08.12.553095. doi: 10.1101/2023.08.12.553095.
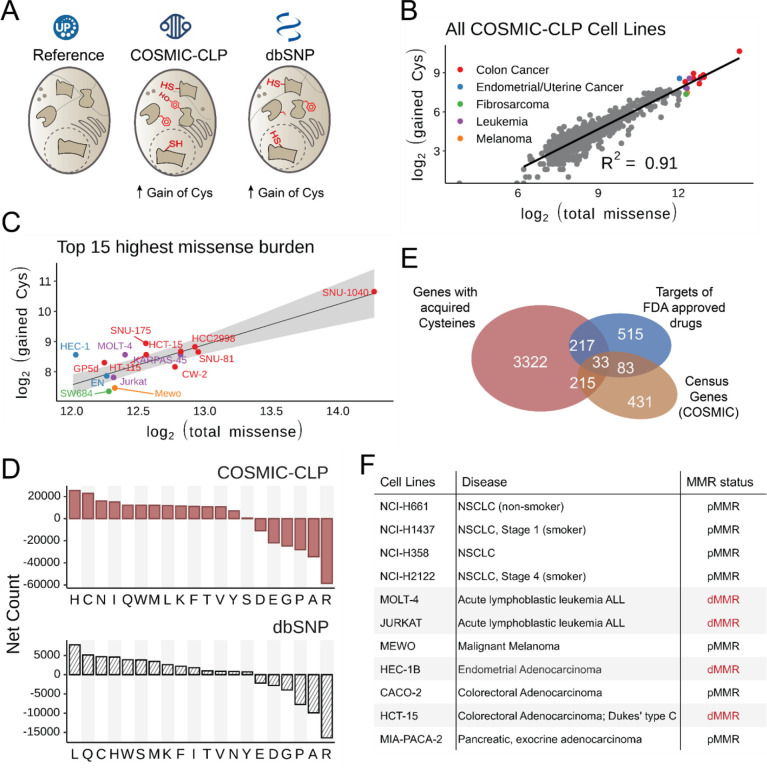
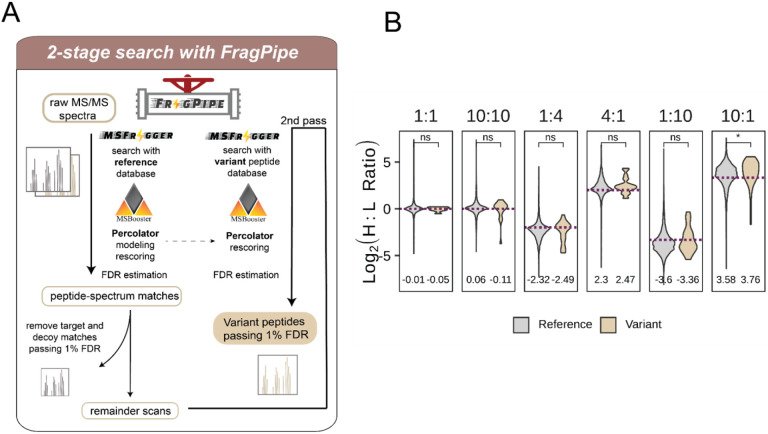
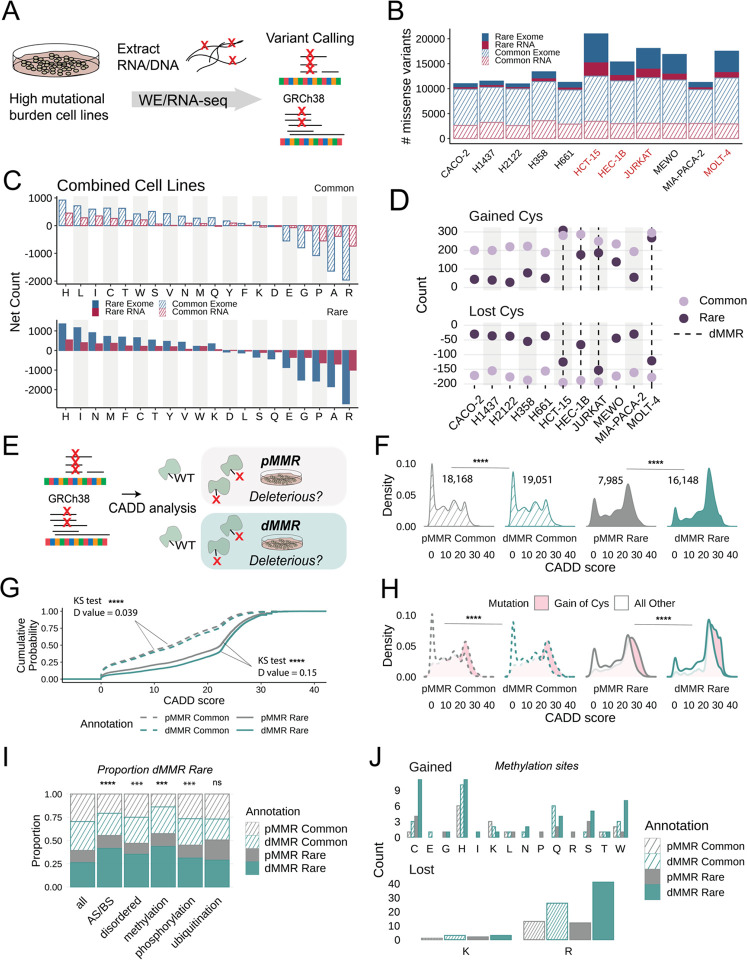
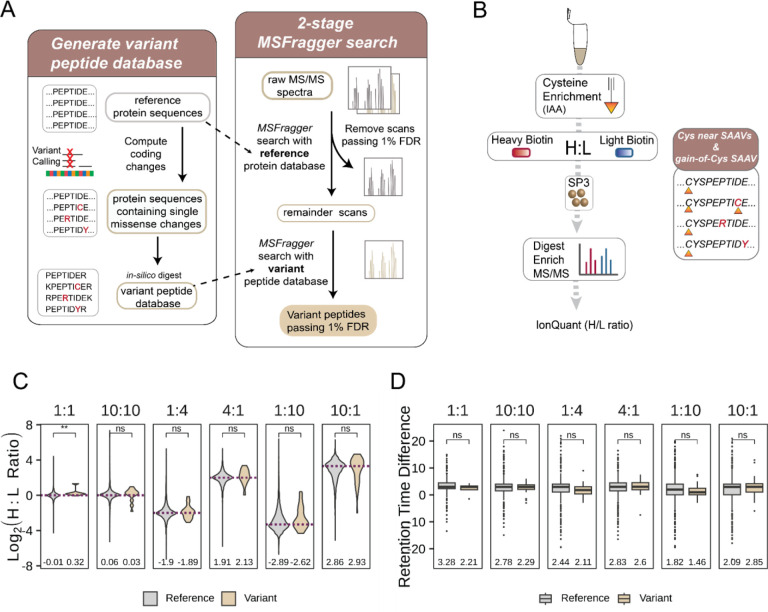
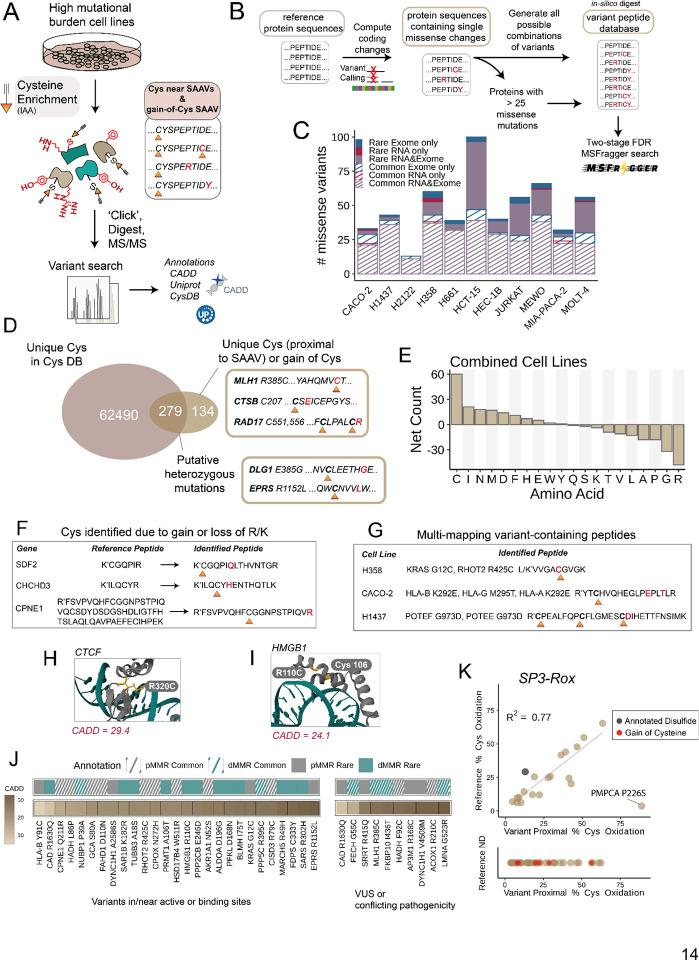
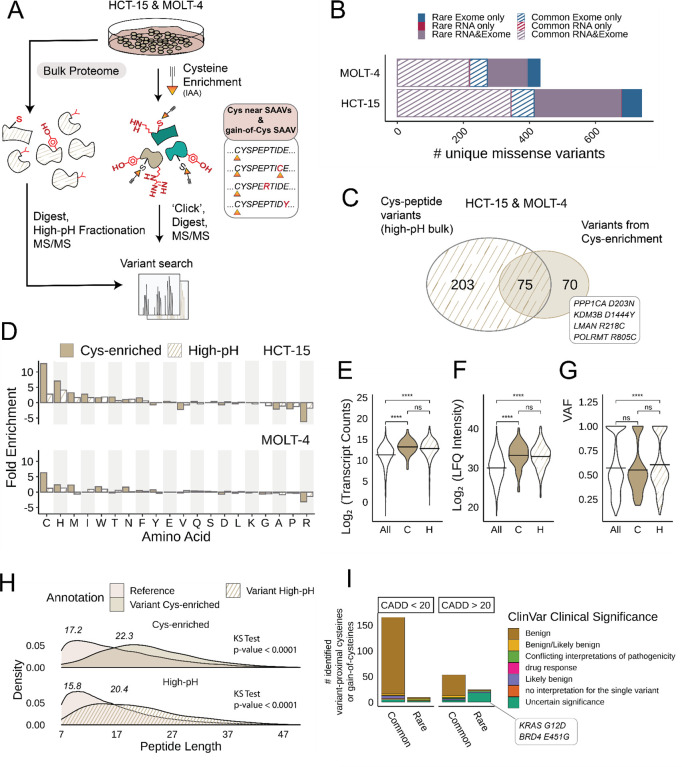
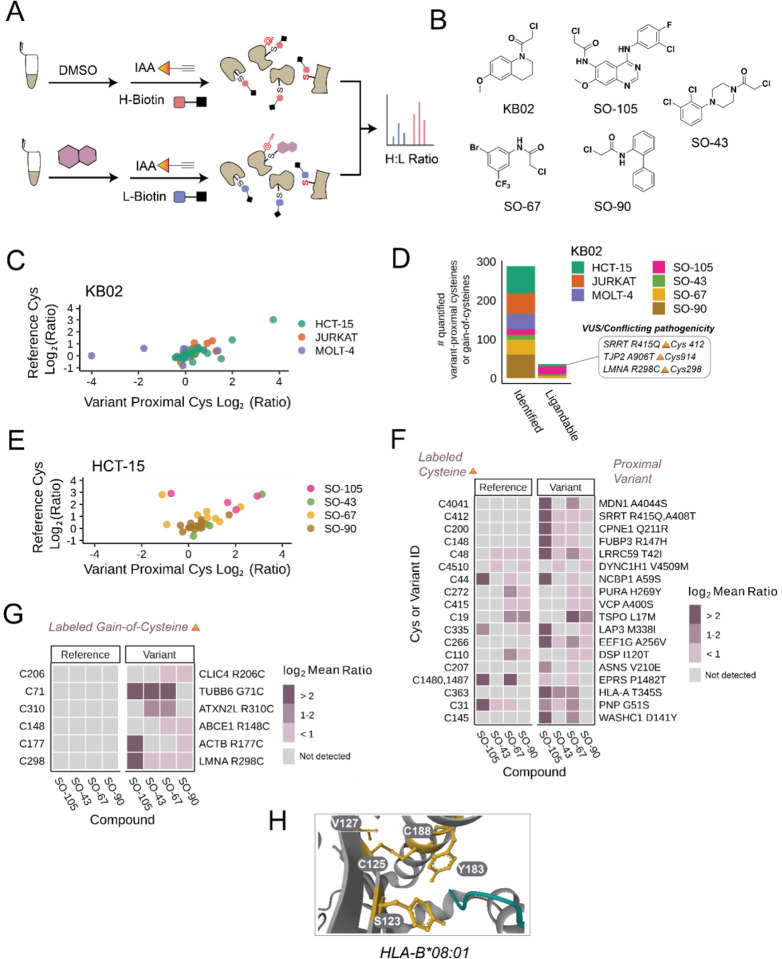
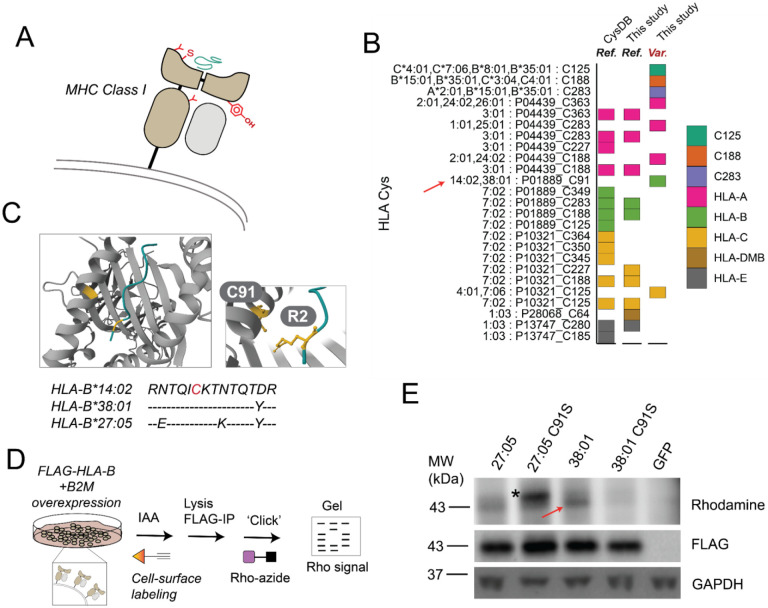
Cancer genomes are rife with genetic variants; one key outcome of this variation is gain-ofcysteine, which is the most frequently acquired amino acid due to missense variants in COSMIC. Acquired cysteines are both driver mutations and sites targeted by precision therapies. However, despite their ubiquity, nearly all acquired cysteines remain uncharacterized. Here, we pair cysteine chemoproteomics-a technique that enables proteome-wide pinpointing of functional, redox sensitive, and potentially druggable residues-with genomics to reveal the hidden landscape of cysteine acquisition. For both cancer and healthy genomes, we find that cysteine acquisition is a ubiquitous consequence of genetic variation that is further elevated in the context of decreased DNA repair. Our chemoproteogenomics platform integrates chemoproteomic, whole exome, and RNA-seq data, with a customized 2-stage false discovery rate (FDR) error controlled proteomic search, further enhanced with a user-friendly FragPipe interface. Integration of CADD predictions of deleteriousness revealed marked enrichment for likely damaging variants that result in acquisition of cysteine. By deploying chemoproteogenomics across eleven cell lines, we identify 116 gain-of-cysteines, of which 10 were liganded by electrophilic druglike molecules. Reference cysteines proximal to missense variants were also found to be pervasive, 791 in total, supporting heretofore untapped opportunities for proteoform-specific chemical probe development campaigns. As chemoproteogenomics is further distinguished by sample-matched combinatorial variant databases and compatible with redox proteomics and small molecule screening, we expect widespread utility in guiding proteoform-specific biology and therapeutic discovery.
癌症基因组中充斥着基因变异;这种变异的一个关键结果是半胱氨酸获得,这是由于COSMIC中的错义变异而最常获得的氨基酸。获得性半胱氨酸既是驱动突变,也是精准治疗的靶点。然而,尽管它们普遍存在,但几乎所有获得性半胱氨酸仍未得到表征。在这里,我们将半胱氨酸化学蛋白质组学(一种能够在全蛋白质组范围内精确识别功能性、氧化还原敏感且可能可成药的残基的技术)与基因组学相结合,以揭示半胱氨酸获得的隐藏情况。对于癌症基因组和健康基因组,我们发现半胱氨酸获得是基因变异的普遍结果,在DNA修复减少的情况下会进一步增加。我们的化学蛋白质基因组学平台整合了化学蛋白质组学、全外显子组和RNA测序数据,通过定制的两阶段错误发现率(FDR)误差控制蛋白质组搜索,并通过用户友好的FragPipe界面进一步增强。整合CADD有害性预测揭示了导致半胱氨酸获得的可能有害变异的显著富集。通过在11个细胞系中部署化学蛋白质基因组学,我们鉴定出了新的116个半胱氨酸获得位点,其中10个被亲电类药物分子配体化。在错义变异附近的参考半胱氨酸也很普遍,总共发现了791个,这为蛋白质异构体特异性化学探针开发活动提供了前所未有的机会。由于化学蛋白质基因组学的进一步特点是样本匹配的组合变异数据库,并与氧化还原蛋白质组学和小分子筛选兼容,我们预计它在指导蛋白质异构体特异性生物学和治疗发现方面具有广泛的应用。