Shanghai Stomatological Hospital & School of Stomatology, State Key Lab of Genetic Engineering, MOE Engineering Research Center of Gene Technology, Shanghai Engineering Research Center of Industrial Microorganisms, School of Life Sciences, Fudan University, Shanghai, 200438, PR China.
Fudan University Shanghai Cancer Center and Department of Pathology, Shanghai Medical College, Fudan University, Shanghai, 200032, PR China.
Redox Biol. 2023 Nov;67:102872. doi: 10.1016/j.redox.2023.102872. Epub 2023 Sep 5.
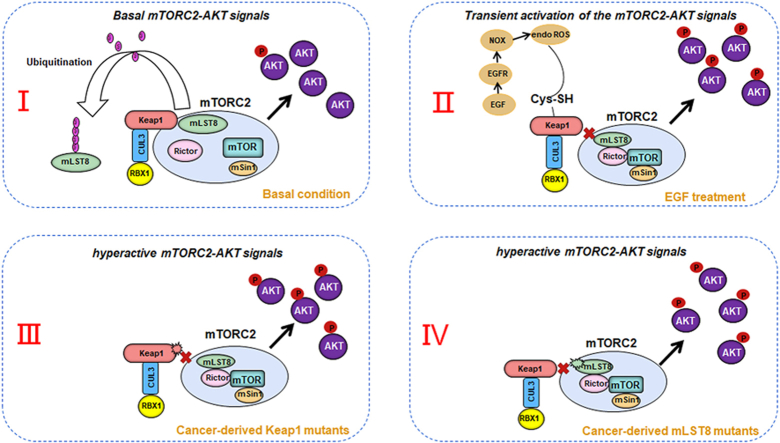
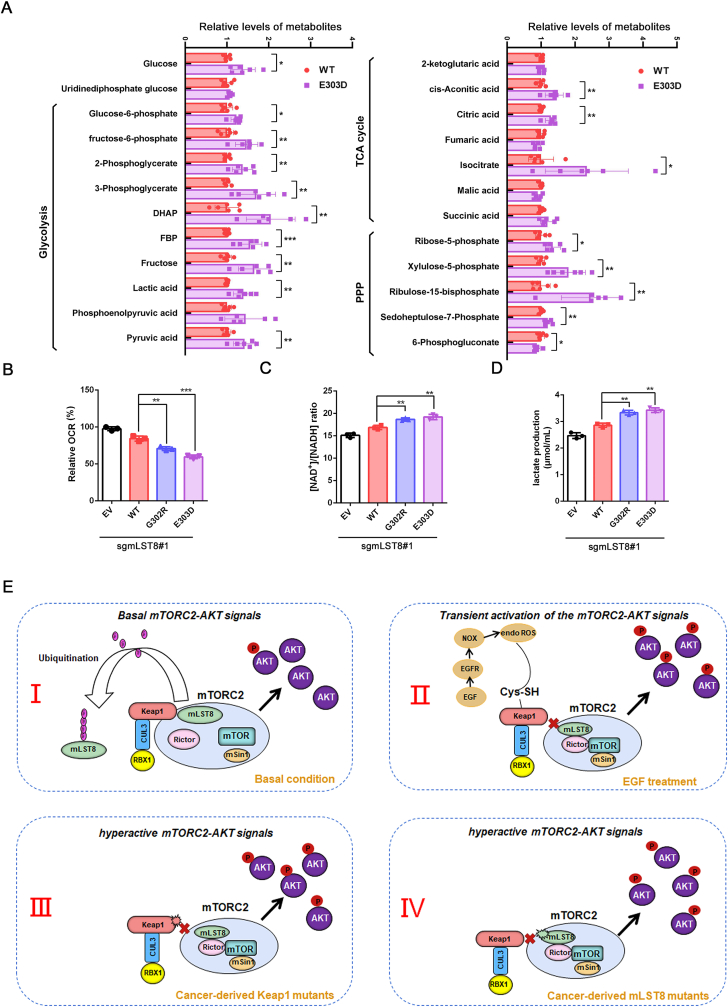
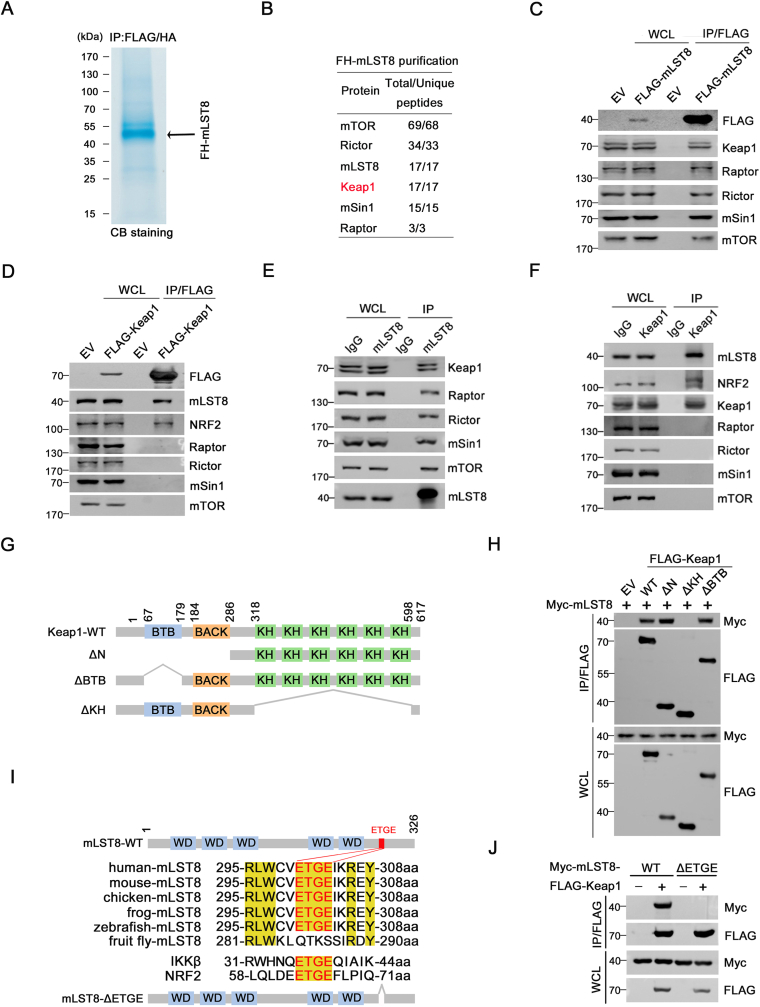
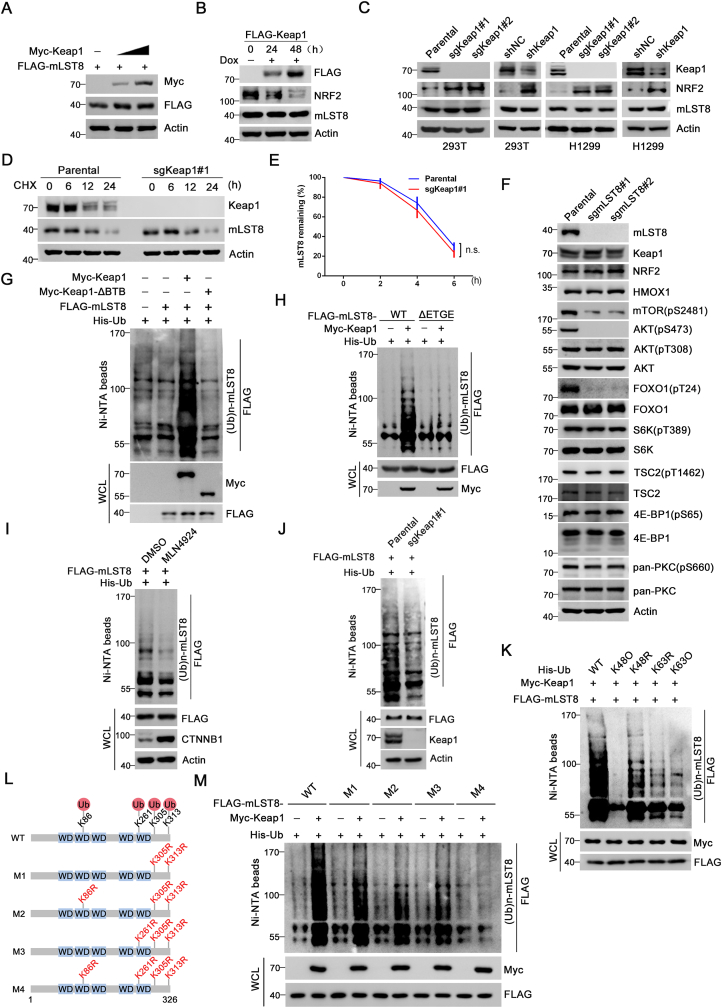
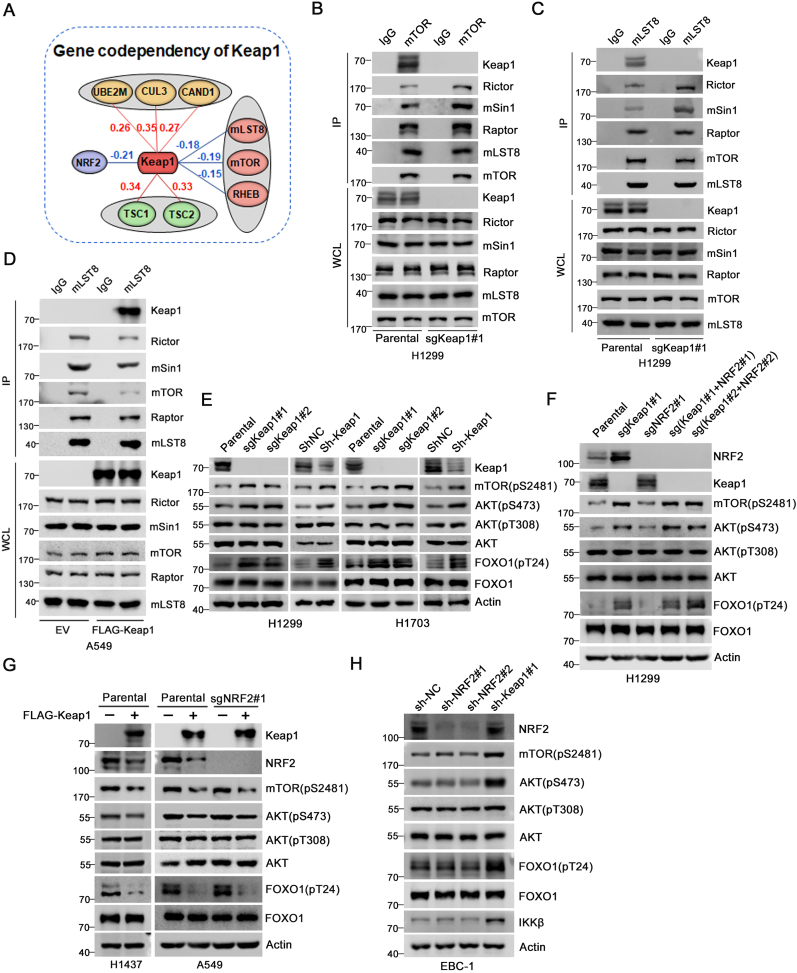
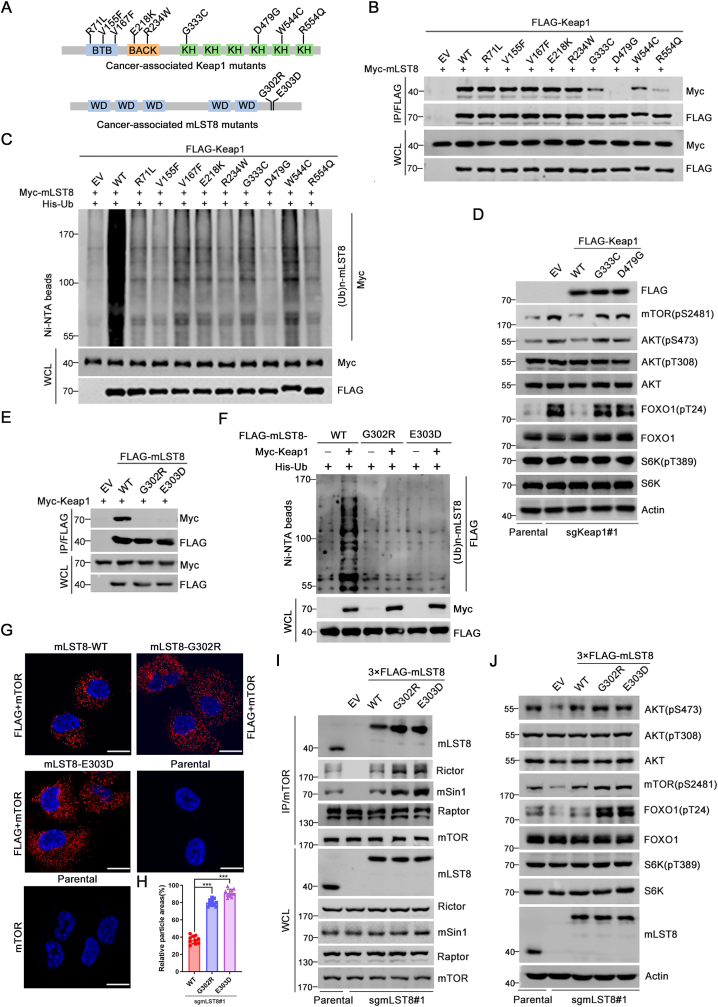
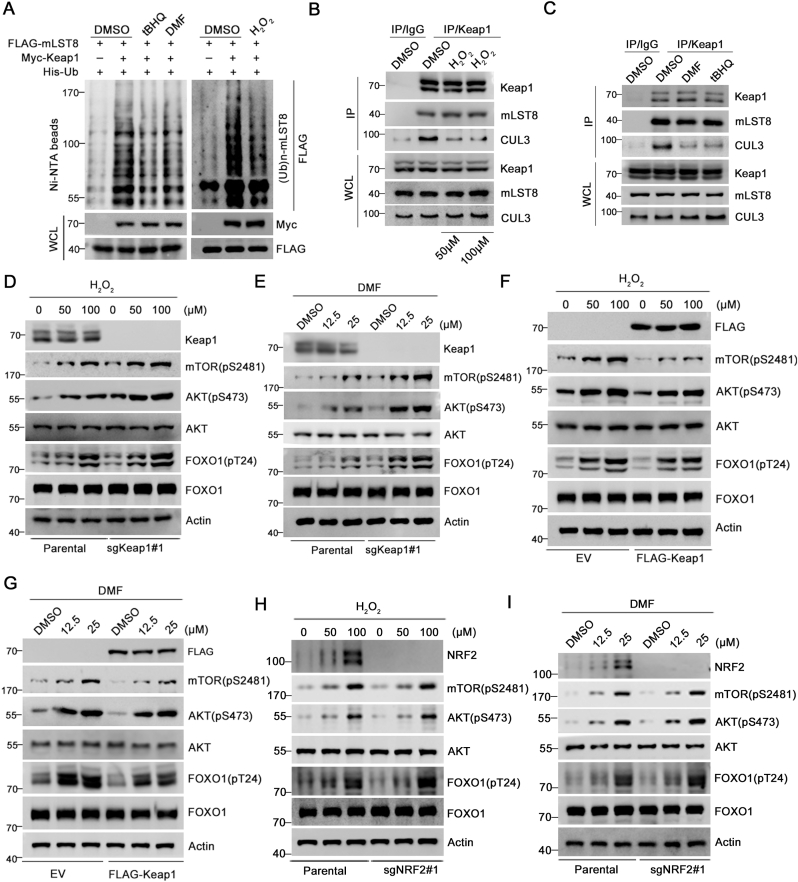
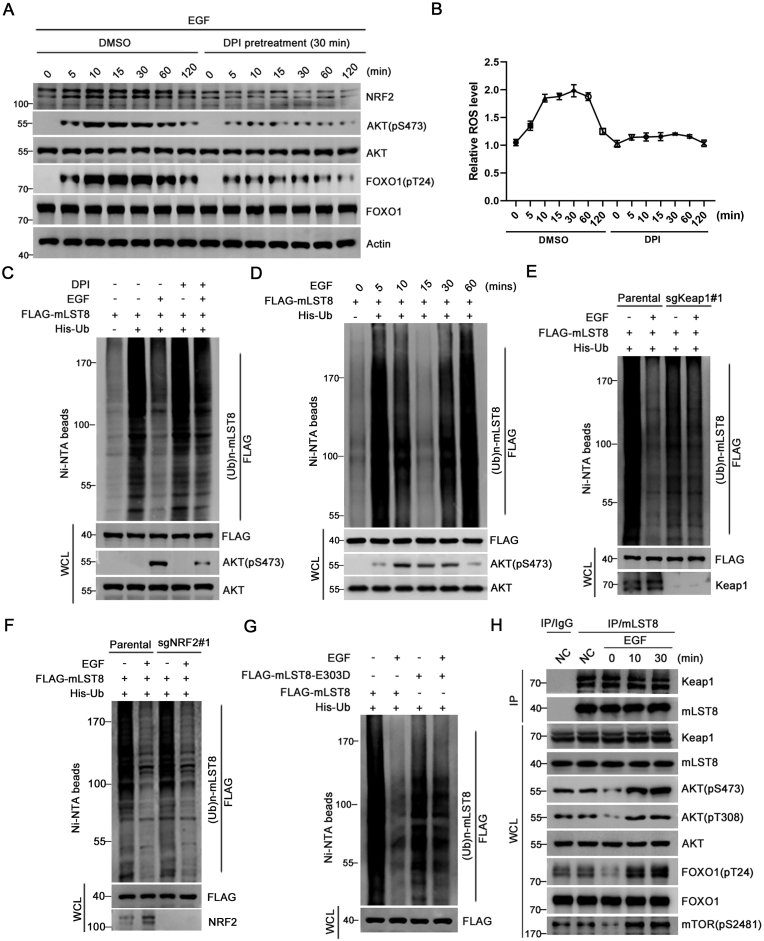
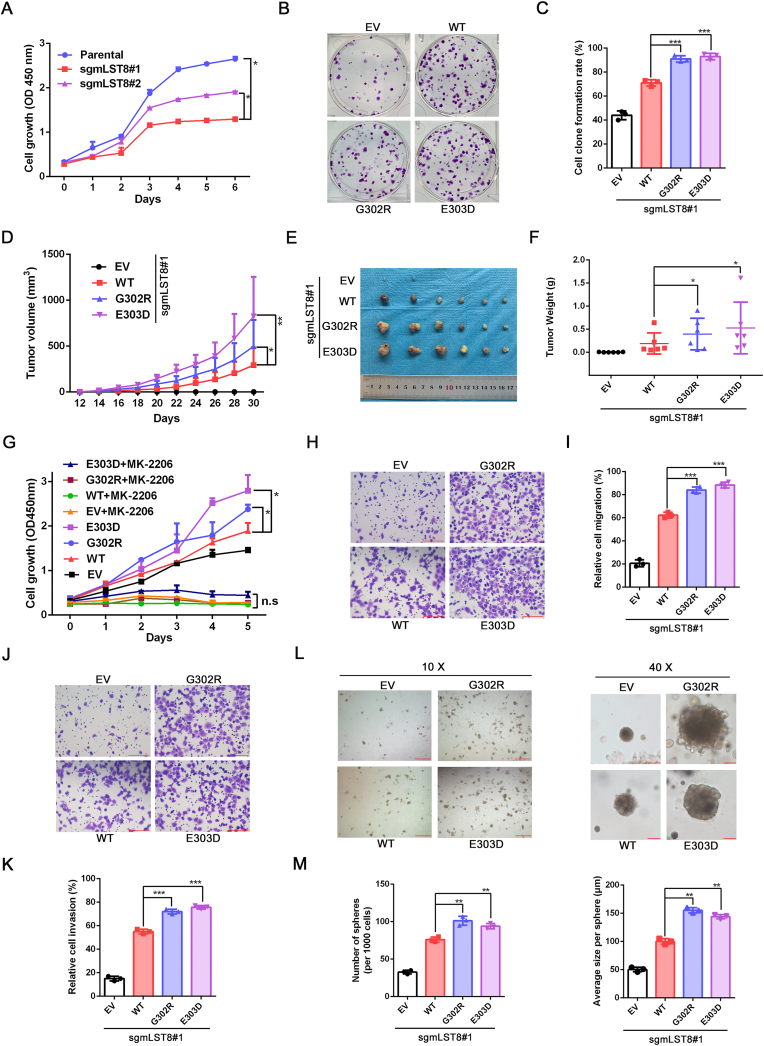
The mechanistic target of the rapamycin (mTOR) pathway, which participates in the regulation of cellular growth and metabolism, is aberrantly regulated in various cancer types. The mTOR complex 2 (mTORC2), which consists of the core components mTOR, Rictor, mSin1, and mLST8, primarily responds to growth signals. However, the coordination between mTORC2 assembly and activity remains poorly understood. Keap1, a major sensor of oxidative stress in cells, functions as a substrate adaptor for Cullin 3-RING E3 ubiquitin ligase (CRL3) to promote proteasomal degradation of NF-E2-related factor 2 (NRF2), which is a transcription factor that protects cells against oxidative and electrophilic stress. In the present study, we demonstrate that Keap1 binds to mLST8 via a conserved ETGE motif. The CRL3 ubiquitin ligase complex promotes non-degradative ubiquitination of mLST8, thus reducing mTORC2 complex integrity and mTORC2-AKT activation. However, this effect can be prevented by oxidative/electrophilic stresses and growth factor signaling-induced reactive oxygen species (ROS) burst. Cancer-derived Keap1 or mLST8 mutations disrupt the Keap1-mLST8 interaction and allow mLST8 to evade Keap1-mediated ubiquitination, thereby enhancing mTORC2-AKT activation and promoting cell malignancy and remodeling cell metabolism. Our findings provide new insights into the molecular mechanisms of Keap1/mLST8 mutation-driven tumorigenesis by promoting mTORC2-AKT activation, which is independent of the canonical NRF2 pathway.
雷帕霉素(mTOR)途径的作用靶点参与细胞生长和代谢的调节,在各种癌症类型中失调。由 mTOR、Rictor、mSin1 和 mLST8 等核心成分组成的 mTOR 复合物 2(mTORC2)主要对生长信号做出反应。然而,mTORC2 组装和活性之间的协调仍然知之甚少。Keap1 是细胞中氧化应激的主要传感器,作为 Cullin 3-RING E3 泛素连接酶(CRL3)的底物衔接子,促进 NF-E2 相关因子 2(NRF2)的蛋白酶体降解,NRF2 是一种转录因子,可保护细胞免受氧化和亲电应激。在本研究中,我们证明 Keap1 通过保守的 ETGE 基序与 mLST8 结合。CRL3 泛素连接酶复合物促进 mLST8 的非降解泛素化,从而降低 mTORC2 复合物的完整性和 mTORC2-AKT 的激活。然而,这种效应可以被氧化/亲电应激和生长因子信号诱导的活性氧(ROS)爆发所阻止。致癌性 Keap1 或 mLST8 突变破坏了 Keap1-mLST8 相互作用,使 mLST8 逃避 Keap1 介导的泛素化,从而增强 mTORC2-AKT 的激活,并促进细胞恶性转化和重塑细胞代谢。我们的研究结果提供了新的见解,即通过促进 mTORC2-AKT 的激活,Keap1/mLST8 突变驱动肿瘤发生的分子机制独立于经典的 NRF2 途径。