Departments of Bioengineering, Biology, Chemistry and Chemical Biology, Single Cell Proteomics Center, Northeastern University, Boston, Massachusetts 02115, United States.
Parallel Squared Technology Institute, Watertown, Massachusetts 02472, United States.
J Proteome Res. 2023 Oct 6;22(10):3149-3158. doi: 10.1021/acs.jproteome.3c00177. Epub 2023 Sep 11.
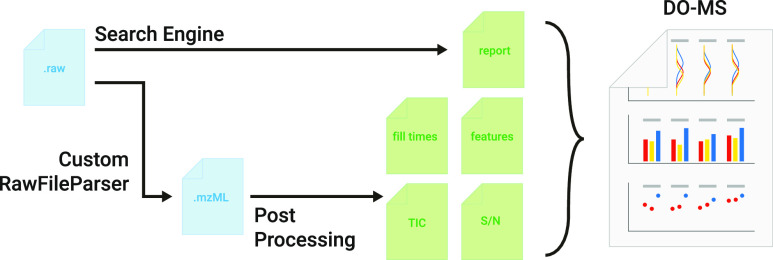
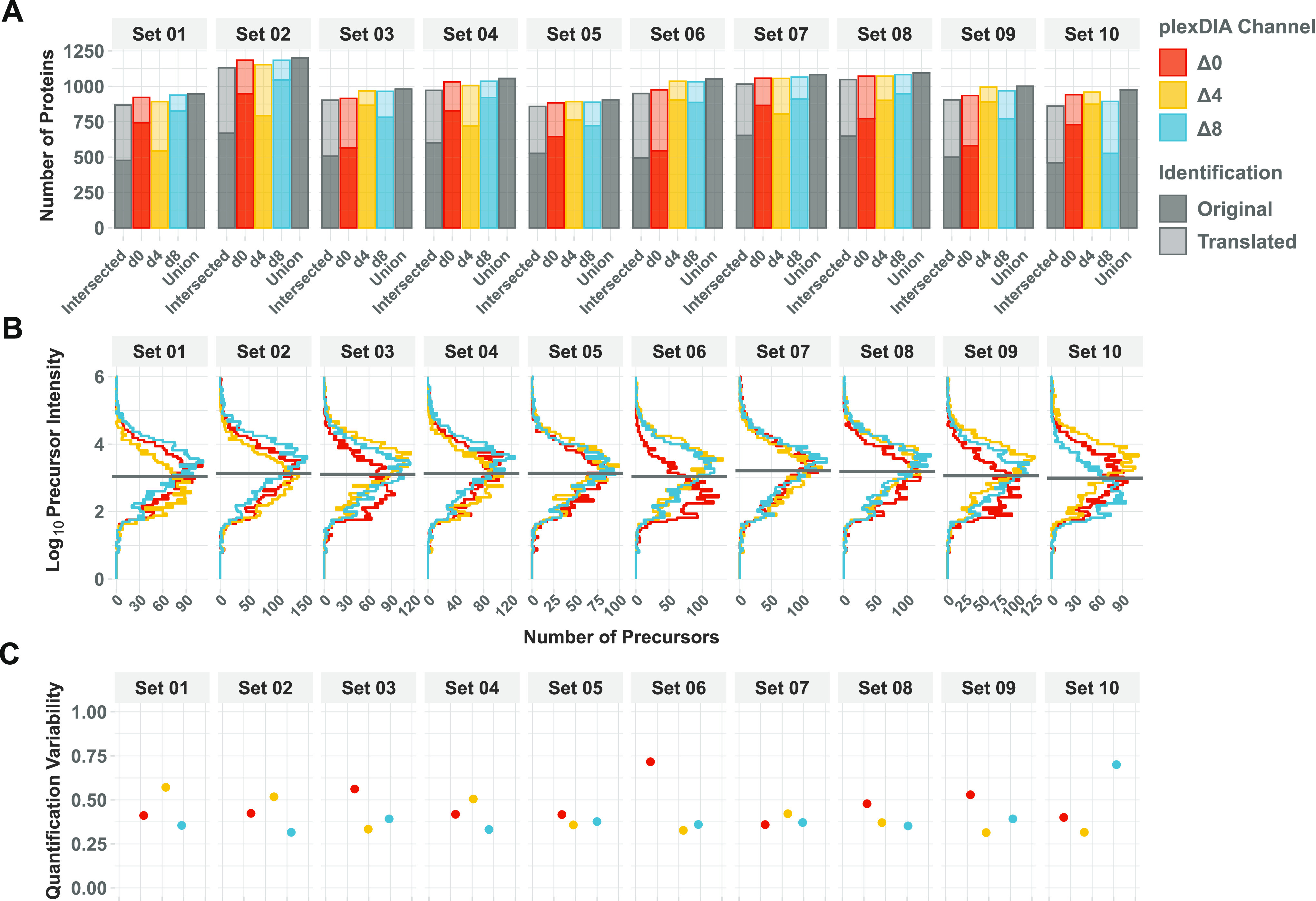
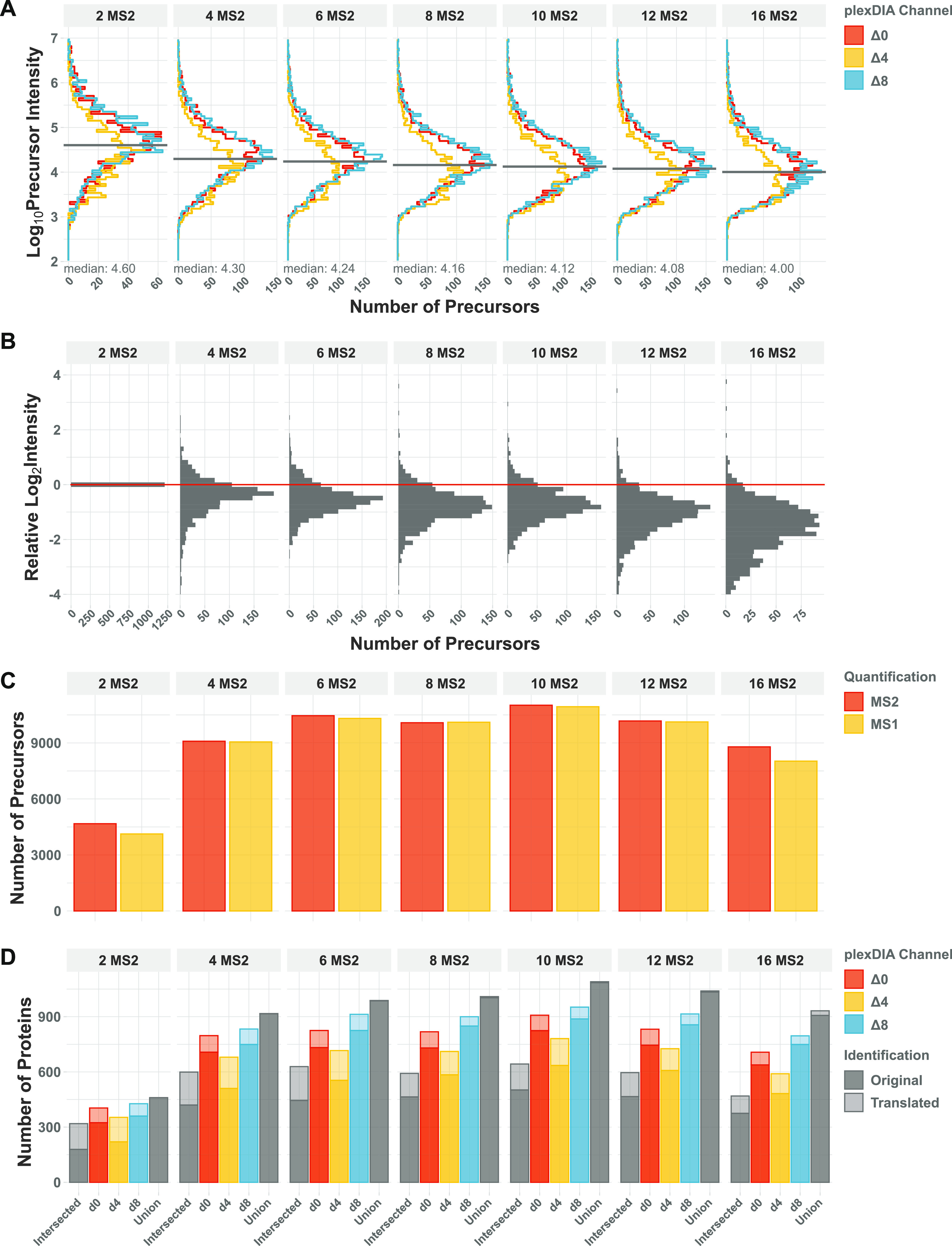
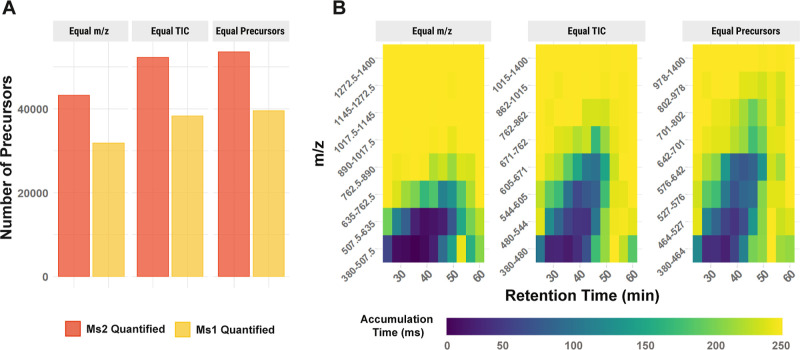
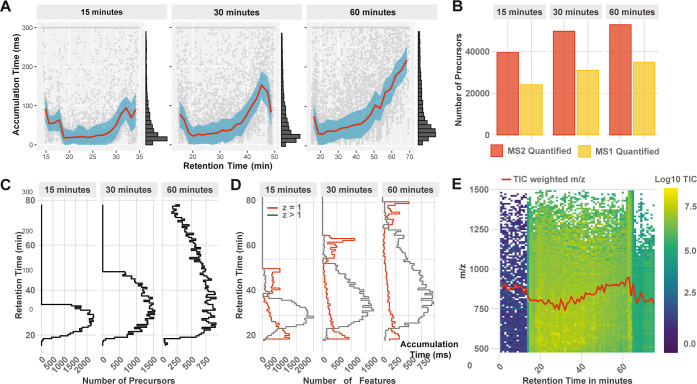
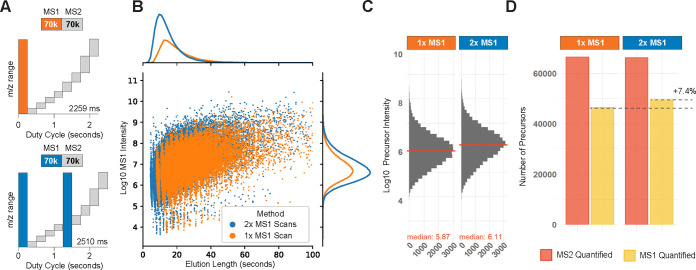
Mass spectrometry (MS) enables specific and accurate quantification of proteins with ever-increasing throughput and sensitivity. Maximizing this potential of MS requires optimizing data acquisition parameters and performing efficient quality control for large datasets. To facilitate these objectives for data-independent acquisition (DIA), we developed a second version of our framework for data-driven optimization of MS methods (DO-MS). The DO-MS app v2.0 (do-ms.slavovlab.net) allows one to optimize and evaluate results from both label-free and multiplexed DIA (plexDIA) and supports optimizations particularly relevant to single-cell proteomics. We demonstrate multiple use cases, including optimization of duty cycle methods, peptide separation, number of survey scans per duty cycle, and quality control of single-cell plexDIA data. DO-MS allows for interactive data display and generation of extensive reports, including publication of quality figures that can be easily shared. The source code is available at github.com/SlavovLab/DO-MS.
质谱(MS)能够特异性和准确地定量蛋白质,具有越来越高的通量和灵敏度。为了最大限度地发挥 MS 的潜力,需要优化数据采集参数,并对大型数据集进行有效的质量控制。为了促进数据非依赖性采集(DIA)的这些目标,我们开发了我们的用于 MS 方法数据驱动优化(DO-MS)框架的第二个版本。DO-MS app v2.0(do-ms.slavovlab.net)允许对无标记和多路复用 DIA(plexDIA)进行优化和评估结果,并支持对单细胞蛋白质组学特别相关的优化。我们展示了多个用例,包括工作循环方法、肽分离、每个工作循环的总扫描次数和单细胞 plexDIA 数据的质量控制的优化。DO-MS 允许交互式数据显示和生成广泛的报告,包括易于共享的质量图的发布。源代码可在 github.com/SlavovLab/DO-MS 获得。