Division of Biomedical Informatics, Department of Medicine, University of California San Diego, La Jolla, CA 92093, United States.
Department of Mathematics, University of California San Diego, La Jolla, CA 92093, United States.
Bioinformatics. 2023 Sep 2;39(9). doi: 10.1093/bioinformatics/btad561.
While evolutionary approaches to medicine show promise, measuring evolution itself is difficult due to experimental constraints and the dynamic nature of body systems. In cancer evolution, continuous observation of clonal architecture is impossible, and longitudinal samples from multiple timepoints are rare. Increasingly available DNA sequencing datasets at single-cell resolution enable the reconstruction of past evolution using mutational history, allowing for a better understanding of dynamics prior to detectable disease. There is an unmet need for an accurate, fast, and easy-to-use method to quantify clone growth dynamics from these datasets.
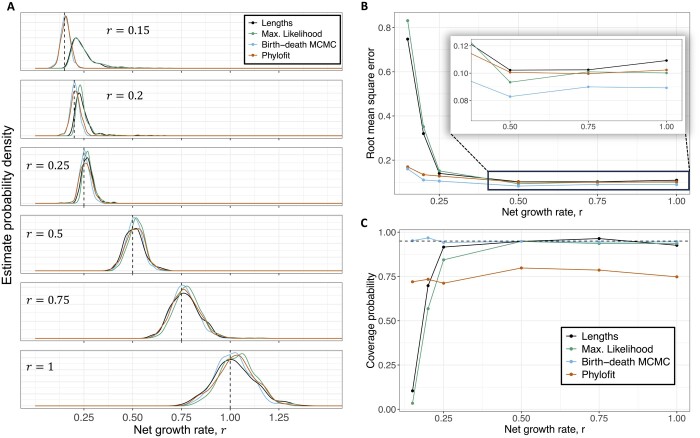
We derived methods based on coalescent theory for estimating the net growth rate of clones using either reconstructed phylogenies or the number of shared mutations. We applied and validated our analytical methods for estimating the net growth rate of clones, eliminating the need for complex simulations used in previous methods. When applied to hematopoietic data, we show that our estimates may have broad applications to improve mechanistic understanding and prognostic ability. Compared to clones with a single or unknown driver mutation, clones with multiple drivers have significantly increased growth rates (median 0.94 versus 0.25 per year; P = 1.6×10-6). Further, stratifying patients with a myeloproliferative neoplasm (MPN) by the growth rate of their fittest clone shows that higher growth rates are associated with shorter time to MPN diagnosis (median 13.9 versus 26.4 months; P = 0.0026).
We developed a publicly available R package, cloneRate, to implement our methods (Package website: https://bdj34.github.io/cloneRate/). Source code: https://github.com/bdj34/cloneRate/.
尽管基于进化的医学方法显示出了前景,但由于实验限制和身体系统的动态性质,测量进化本身具有一定难度。在癌症进化中,对克隆结构的连续观察是不可能的,而且很少有来自多个时间点的纵向样本。越来越多的单细胞分辨率 DNA 测序数据集使我们能够使用突变历史重建过去的进化,从而更好地了解可检测疾病之前的动态。目前迫切需要一种准确、快速且易于使用的方法,以便能够从这些数据集定量克隆生长动态。
我们基于合并理论推导出了一些方法,这些方法可用于使用重建的系统发育或共享突变的数量来估计克隆的净增长率。我们应用并验证了我们用于估计克隆净增长率的分析方法,从而消除了以前方法中使用的复杂模拟。当应用于造血数据时,我们表明我们的估计值可能具有广泛的应用,可用于改善对机制的理解和预后能力。与具有单个或未知驱动突变的克隆相比,具有多个驱动突变的克隆的增长率明显更高(中位数分别为每年 0.94 与 0.25;P=1.6×10-6)。此外,根据最适合克隆的增长率对患有骨髓增殖性肿瘤(MPN)的患者进行分层,表明更高的增长率与 MPN 诊断时间更短相关(中位数分别为 13.9 与 26.4 个月;P=0.0026)。
我们开发了一个可公开访问的 R 包 cloneRate 来实现我们的方法(包网站:https://bdj34.github.io/cloneRate/)。源代码:https://github.com/bdj34/cloneRate/。