Gonzales Emmanuel, Gardin Antoine, Almes Marion, Darmellah-Remil Amaria, Seguin Hanh, Mussini Charlotte, Franchi-Abella Stéphanie, Duché Mathieu, Ackermann Oanez, Thébaut Alice, Habes Dalila, Hermeziu Bogdan, Lapalus Martine, Falguières Thomas, Combal Jean-Philippe, Benichou Bernard, Valero Sonia, Davit-Spraul Anne, Jacquemin Emmanuel
Pediatric Hepatology and Liver Transplantation, National Reference Centre for Biliary Atresia and Genetic Cholestasis, FILFOIE, ERN RARE LIVER, France.
Inserm U1193, Hepatinov, University Paris-Saclay, Orsay, France.
JHEP Rep. 2023 Jul 13;5(10):100844. doi: 10.1016/j.jhepr.2023.100844. eCollection 2023 Oct.
BACKGROUND & AIMS: Progressive familial intrahepatic cholestasis type 3 (PFIC3) is a rare liver disease caused by biallelic variations in . Data reporting on the impact of genotype and of response to ursodeoxycholic acid (UDCA) therapy on long-term outcomes are scarce.
We retrospectively describe a cohort of 38 patients with PFIC3 with a median age at last follow-up of 19.5 years (range 3.8-53.8).
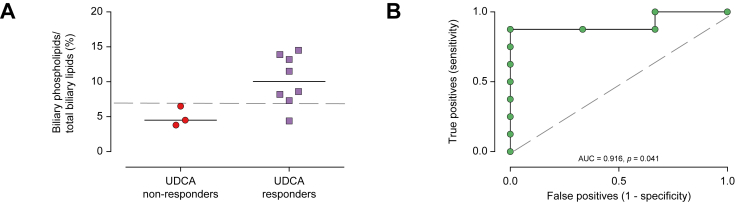
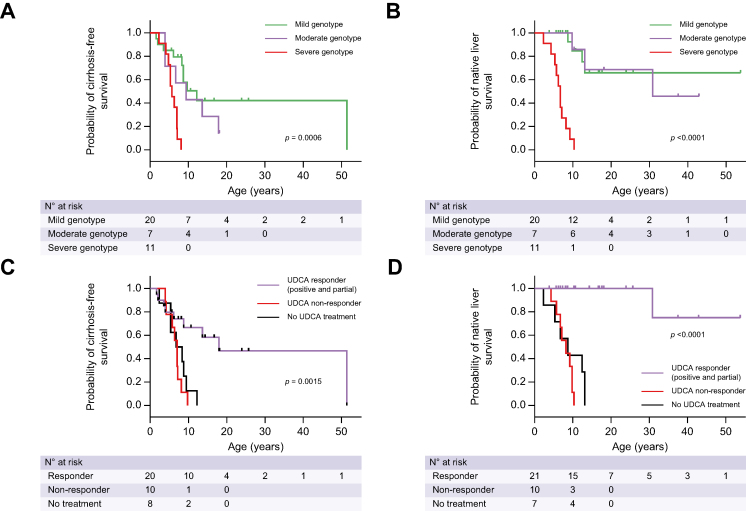
Twenty patients presented with symptoms before 1 year of age. Thirty-one patients received ursodeoxycholic acid (UDCA) therapy resulting in serum liver test improvement in 20. Twenty-seven patients had cirrhosis at a median age of 8.1 years of whom 18 received a liver transplant at a median age of 8.5 years. Patients carrying at least one missense variation were more likely to present with positive (normal or decreased) canalicular MDR3 expression in the native liver and had prolonged native liver survival (NLS; median 12.4 years [range 3.8-53.8]). In contrast, in patients with severe genotypes (no missense variation), there was no detectable canalicular MDR3 expression, symptom onset and cirrhosis occurred earlier, and all underwent liver transplantation (at a median age of 6.7 years [range 2.3-10.3]). The latter group was refractory to UDCA treatment, whereas 87% of patients with at least one missense variation displayed an improvement in liver biochemistry in response to UDCA. Biliary phospholipid levels over 6.9% of total biliary lipid levels predicted response to UDCA. Response to UDCA predicted NLS.
Patients carrying at least one missense variation, with positive canalicular expression of MDR3 and a biliary phospholipid level over 6.9% of total biliary lipid levels were more likely to respond to UDCA and to exhibit prolonged NLS.
In this study, data show that genotype and response to ursodeoxycholic acid therapy predicted native liver survival in patients with PFIC3 (progressive familial intrahepatic cholestasis type 3). Patients carrying at least one missense variation, with positive (decreased or normal) immuno-staining for canalicular MDR3, and a biliary phospholipid level over 6.9% of total biliary lipids were more likely to respond to ursodeoxycholic acid therapy and to exhibit prolonged native liver survival.
3型进行性家族性肝内胆汁淤积症(PFIC3)是一种由[基因名称]双等位基因变异引起的罕见肝脏疾病。关于基因型以及熊去氧胆酸(UDCA)治疗反应对长期预后影响的数据报道较少。
我们回顾性描述了一组38例PFIC3患者,末次随访时的中位年龄为19.5岁(范围3.8 - 53.8岁)。
20例患者在1岁前出现症状。31例患者接受了熊去氧胆酸(UDCA)治疗,其中20例患者的血清肝功能检查有所改善。27例患者发生肝硬化,中位年龄为8.1岁,其中18例在中位年龄8.5岁时接受了肝移植。携带至少一个错义变异的患者,其天然肝脏中胆小管MDR3表达更可能呈阳性(正常或降低),并且天然肝脏生存期(NLS)延长(中位12.4年[范围3.8 - 53.8年])。相比之下,在具有严重基因型(无错义变异)的患者中,未检测到胆小管MDR3表达,症状出现和肝硬化发生更早,并且所有患者均接受了肝移植(中位年龄6.7岁[范围2.3 - 10.3岁])。后一组患者对UDCA治疗无效,而至少携带一个错义变异的患者中,87%对UDCA治疗有肝脏生化指标改善。胆汁磷脂水平超过总胆汁脂质水平的6.9%可预测对UDCA的反应。对UDCA的反应可预测NLS。
携带至少一个错义变异、MDR3胆小管表达呈阳性且胆汁磷脂水平超过总胆汁脂质水平6.9%的患者,更可能对UDCA治疗有反应,并表现出延长的NLS。
在本研究中,数据表明基因型和对熊去氧胆酸治疗的反应可预测PFIC3(3型进行性家族性肝内胆汁淤积症)患者的天然肝脏生存期。携带至少一个错义变异、胆小管MDR3免疫染色呈阳性(降低或正常)且胆汁磷脂水平超过总胆汁脂质6.9%的患者,更可能对熊去氧胆酸治疗有反应,并表现出延长的天然肝脏生存期。