González-Martínez Irene, Cerro-Herreros Estefanía, Moreno Nerea, García-Rey Andrea, Espinosa-Espinosa Jorge, Carrascosa-Sàez Marc, Piqueras-Losilla Diego, Arzumanov Andrey, Seoane-Miraz David, Jad Yahya, Raz Richard, Wood Matthew J, Varela Miguel A, Llamusí Beatriz, Artero Rubén
University Research Institute for Biotechnology and Biomedicine (BIOTECMED), Universidad de Valencia, Valencia, Spain.
Translational Genomics Group, INCLIVA Biomedical Research Institute, Avenue Menéndez Pelayo 4 acc, 46010 Valencia, Spain.
Mol Ther Nucleic Acids. 2023 Sep 5;34:102024. doi: 10.1016/j.omtn.2023.09.001. eCollection 2023 Dec 12.
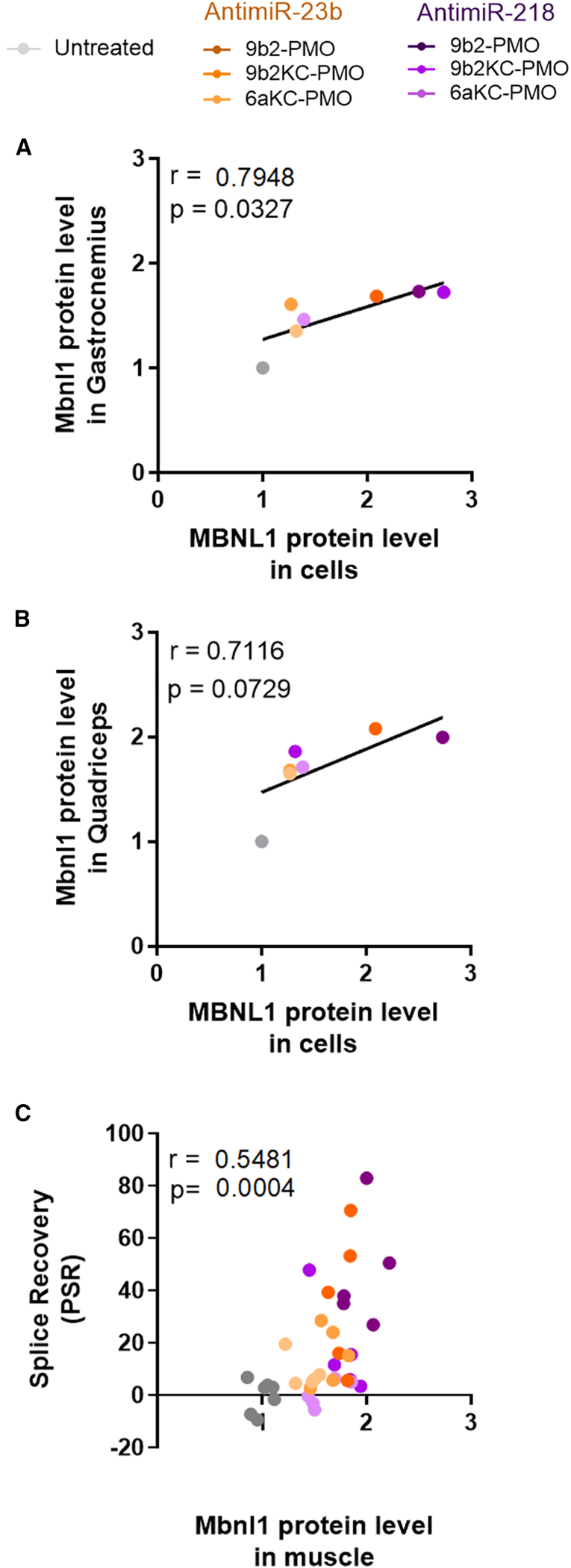
Myotonic dystrophy type 1 (DM1) is a rare neuromuscular disease caused by a CTG repeat expansion in the gene that generates toxic RNA with a myriad of downstream alterations in RNA metabolism. A key consequence is the sequestration of alternative splicing regulatory proteins MBNL1/2 by expanded transcripts in the affected tissues. MBNL1/2 depletion interferes with a developmental alternative splicing switch that causes the expression of fetal isoforms in adults. Boosting the endogenous expression of MBNL proteins by inhibiting the natural translational repressors miR-23b and miR-218 has previously been shown to be a promising therapeutic approach. We designed antimiRs against both miRNAs with a phosphorodiamidate morpholino oligonucleotide (PMO) chemistry conjugated to cell-penetrating peptides (CPPs) to improve delivery to affected tissues. In DM1 cells, CPP-PMOs significantly increased MBNL1 levels. In some candidates, this was achieved using concentrations less than two orders of magnitude below the median toxic concentration, with up to 5.38-fold better therapeutic window than previous antagomiRs. In mice, intravenous injections of CPP-PMOs improve molecular, histopathological, and functional phenotypes, without signs of toxicity. Our findings place CPP-PMOs as promising antimiR candidates to overcome the treatment delivery challenge in DM1 therapy.
1型强直性肌营养不良症(DM1)是一种罕见的神经肌肉疾病,由基因中的CTG重复序列扩增引起,该序列产生有毒的RNA,并在RNA代谢中引发无数下游改变。一个关键后果是,受影响组织中扩增的转录本会隔离剪接调节蛋白MBNL1/2。MBNL1/2的缺失会干扰发育过程中的剪接转换,导致成人中胎儿异构体的表达。此前已证明,通过抑制天然翻译抑制因子miR-23b和miR-218来提高MBNL蛋白的内源性表达是一种很有前景的治疗方法。我们设计了针对这两种miRNA的抗miR,采用与细胞穿透肽(CPP)偶联的磷酰二胺吗啉代寡核苷酸(PMO)化学方法,以改善对受影响组织的递送。在DM1细胞中,CPP-PMO显著提高了MBNL1水平。在一些候选药物中,这是通过使用低于中位毒性浓度两个数量级以下的浓度实现的,其治疗窗口比以前的抗miR高5.38倍。在小鼠中,静脉注射CPP-PMO可改善分子、组织病理学和功能表型,且无毒性迹象。我们的研究结果表明,CPP-PMO是很有前景的抗miR候选药物,可克服DM1治疗中的给药挑战。