Department of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy.
Department of Informatics, Systems and Communication, University of Milano-Bicocca, Milan, Italy.
Nat Commun. 2023 Sep 25;14(1):5982. doi: 10.1038/s41467-023-41670-3.
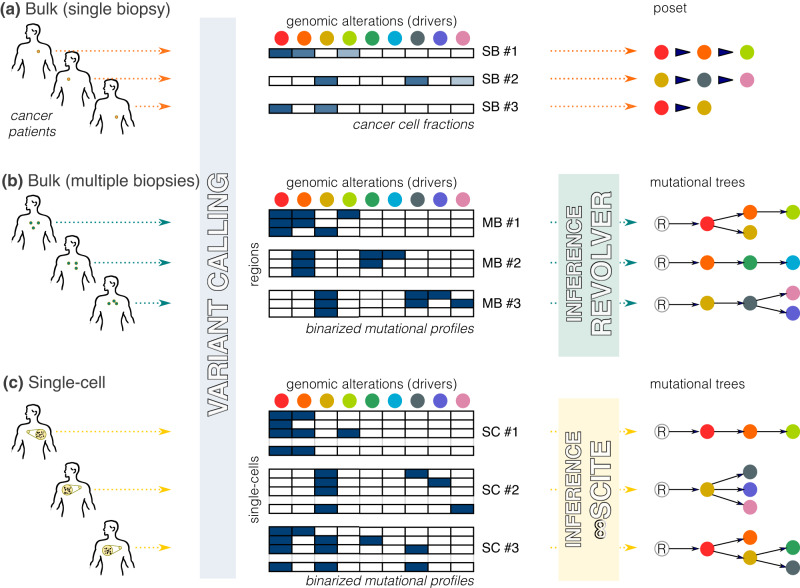
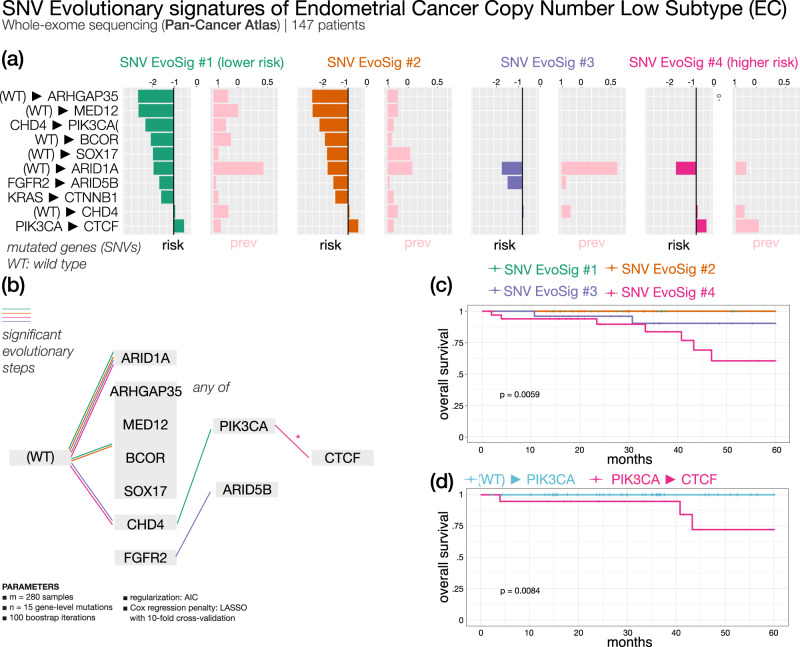
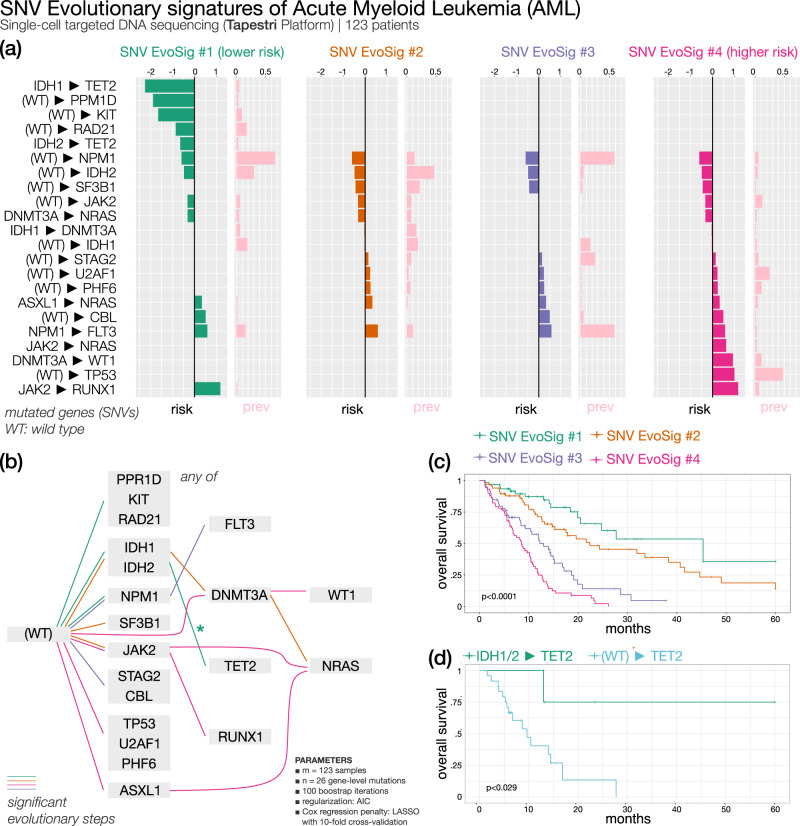
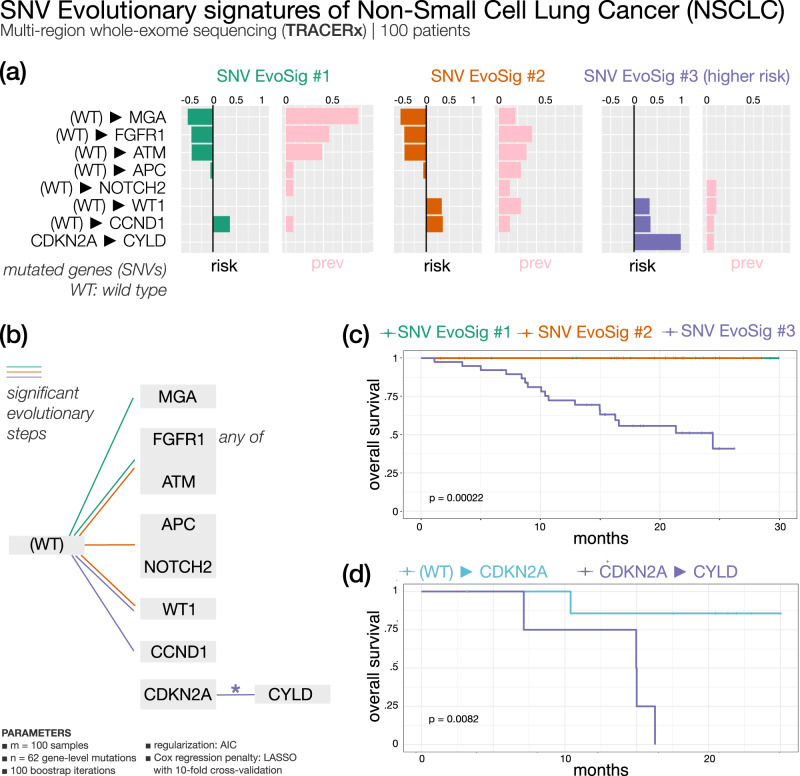
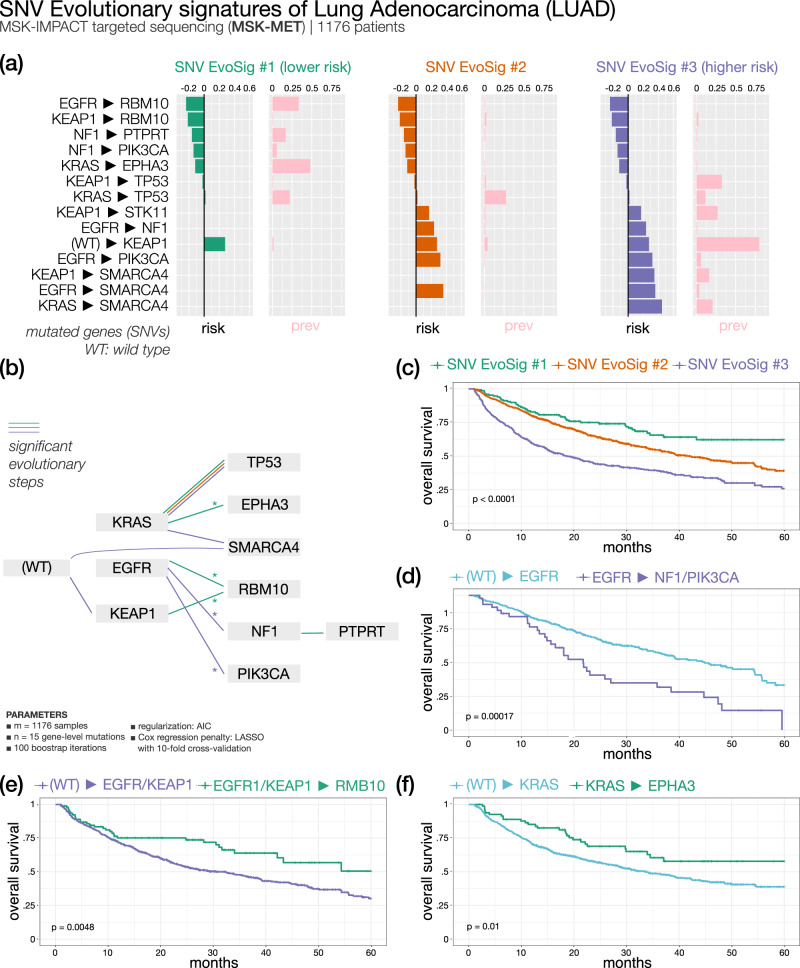
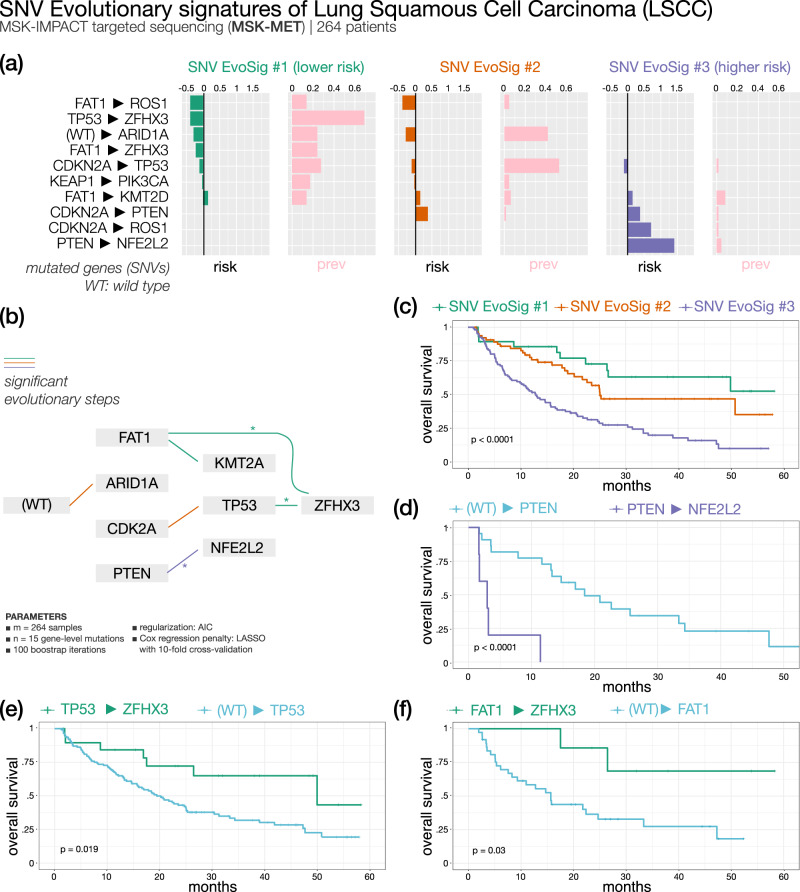
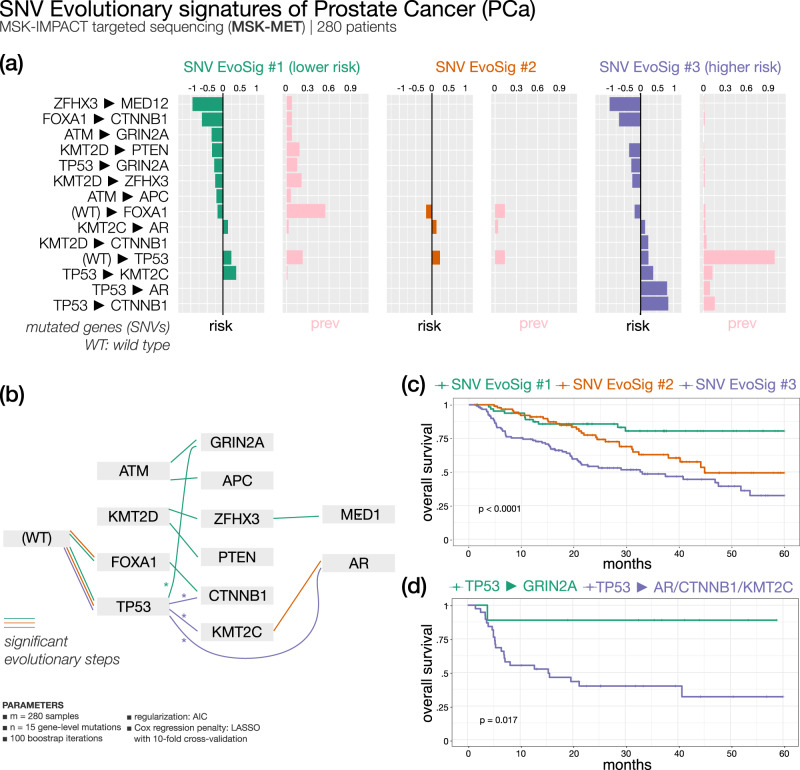
Recurring sequences of genomic alterations occurring across patients can highlight repeated evolutionary processes with significant implications for predicting cancer progression. Leveraging the ever-increasing availability of cancer omics data, here we unveil cancer's evolutionary signatures tied to distinct disease outcomes, representing "favored trajectories" of acquisition of driver mutations detected in patients with similar prognosis. We present a framework named ASCETIC (Agony-baSed Cancer EvoluTion InferenCe) to extract such signatures from sequencing experiments generated by different technologies such as bulk and single-cell sequencing data. We apply ASCETIC to (i) single-cell data from 146 myeloid malignancy patients and bulk sequencing from 366 acute myeloid leukemia patients, (ii) multi-region sequencing from 100 early-stage lung cancer patients, (iii) exome/genome data from 10,000+ Pan-Cancer Atlas samples, and (iv) targeted sequencing from 25,000+ MSK-MET metastatic patients, revealing subtype-specific single-nucleotide variant signatures associated with distinct prognostic clusters. Validations on several datasets underscore the robustness and generalizability of the extracted signatures.
在不同患者中发生的基因组改变的重复序列可以突出具有重要预测癌症进展意义的重复进化过程。利用癌症组学数据的可用性不断增加,我们在这里揭示了与不同疾病结果相关的癌症进化特征,这些特征代表了在具有相似预后的患者中检测到的驱动突变获得的“有利轨迹”。我们提出了一种名为 ASCETIC(基于痛苦的癌症进化推断)的框架,用于从不同技术(如批量和单细胞测序数据)生成的测序实验中提取此类特征。我们将 ASCETIC 应用于:(i) 来自 146 名髓系恶性肿瘤患者的单细胞数据和来自 366 名急性髓系白血病患者的批量测序数据,(ii) 来自 100 名早期肺癌患者的多区域测序数据,(iii) 来自 10,000+ Pan-Cancer Atlas 样本的外显子/基因组数据,以及 (iv) 来自 25,000+ MSK-MET 转移性患者的靶向测序数据,揭示了与不同预后聚类相关的特定于亚型的单核苷酸变异特征。在几个数据集上的验证强调了提取特征的稳健性和通用性。