Lee Seunghoon, Zhai Huanchen, Chan Garnet Kin-Lic
Department of Chemistry, Seoul National University, Seoul 151-747, South Korea.
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States.
J Chem Theory Comput. 2023 Nov 14;19(21):7753-7763. doi: 10.1021/acs.jctc.3c00663. Epub 2023 Oct 18.
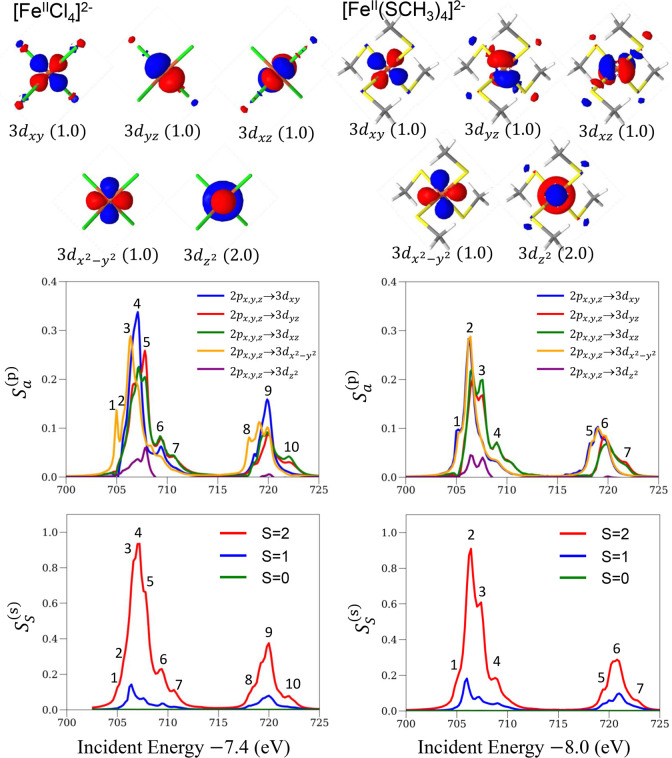
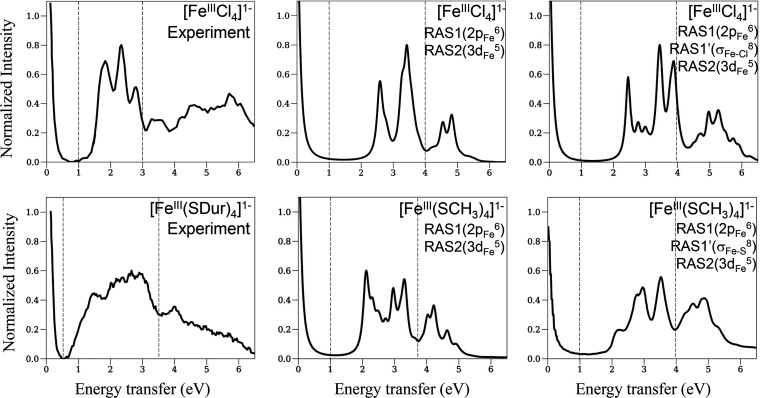
We describe an ab initio approach to simulate L-edge X-ray absorption (XAS) and 2p3d resonant inelastic X-ray scattering (RIXS) spectroscopies. We model the strongly correlated electronic structure within a restricted active space and employ a correction vector formulation instead of sum-over-state expressions for the spectra, thus eliminating the need to calculate a large number of intermediate and final electronic states. We present benchmark simulations of the XAS and RIXS spectra of the iron complexes [FeCl] and [Fe(SCH)] and interpret the spectra by deconvolving the correction vectors. Our approach represents a step toward simulating the X-ray spectroscopies of larger metal cluster systems that play a pivotal role in biology.
我们描述了一种从头算方法来模拟L边X射线吸收(XAS)和2p3d共振非弹性X射线散射(RIXS)光谱。我们在受限活性空间内对强关联电子结构进行建模,并采用校正向量公式而非光谱的态求和表达式,从而无需计算大量的中间和最终电子态。我们给出了铁配合物[FeCl]和[Fe(SCH)]的XAS和RIXS光谱的基准模拟,并通过对校正向量进行去卷积来解释光谱。我们的方法朝着模拟在生物学中起关键作用的更大金属簇系统的X射线光谱迈出了一步。