Xu Guangyang, Liu Hui, Xia Dunling, Zhao Yan, Qian Yao, Han Hongyan, Pan Jiahua, Jiang Hua, Jiang Yongqiang, Sun Gengyun
The First Clinical College of Anhui Medical University, Hefei, China.
State Key Laboratory of Pathogen and Biosecurity, Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences, Beijing, China.
J Thorac Dis. 2023 Sep 28;15(9):4987-5005. doi: 10.21037/jtd-23-1138. Epub 2023 Sep 21.
(SMA) has emerged as an important pathogen capable of causing an opportunistic and nosocomial infection. We performed RNA sequencing (RNA-seq) of lung tissues from mice with pulmonary SMA infection over time via aerosolized intratracheal inhalation to investigate transcription profile changes in SMA-infected lungs.
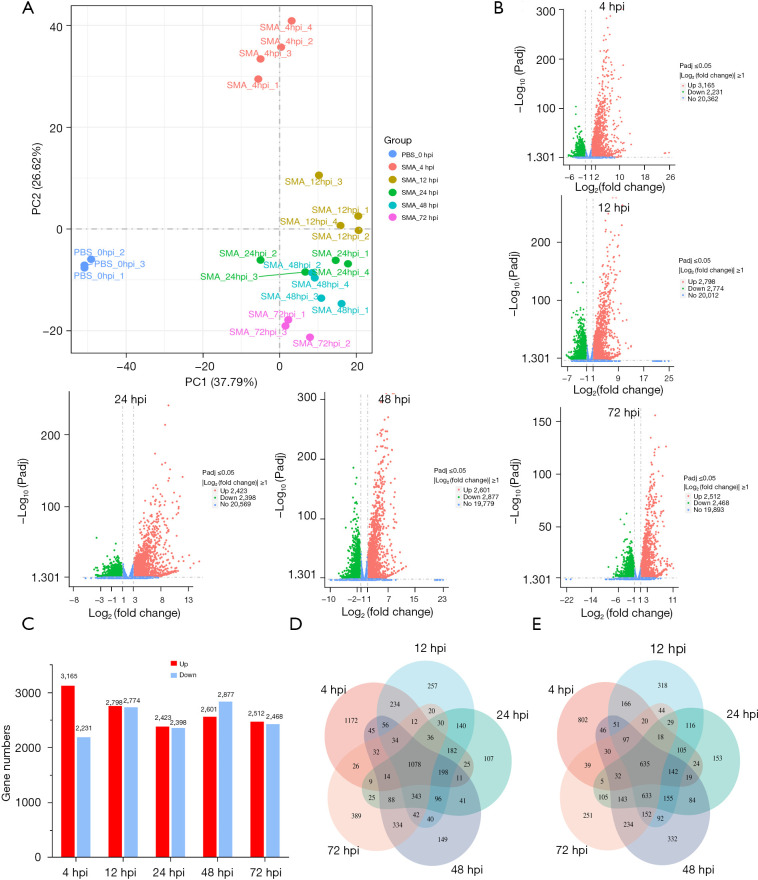
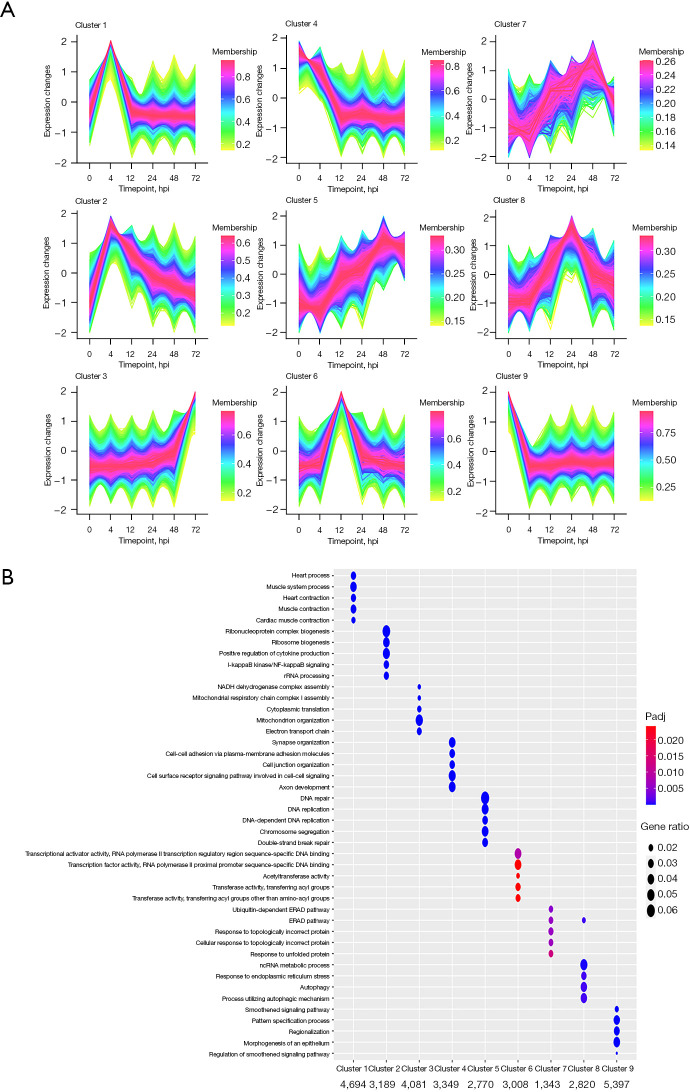
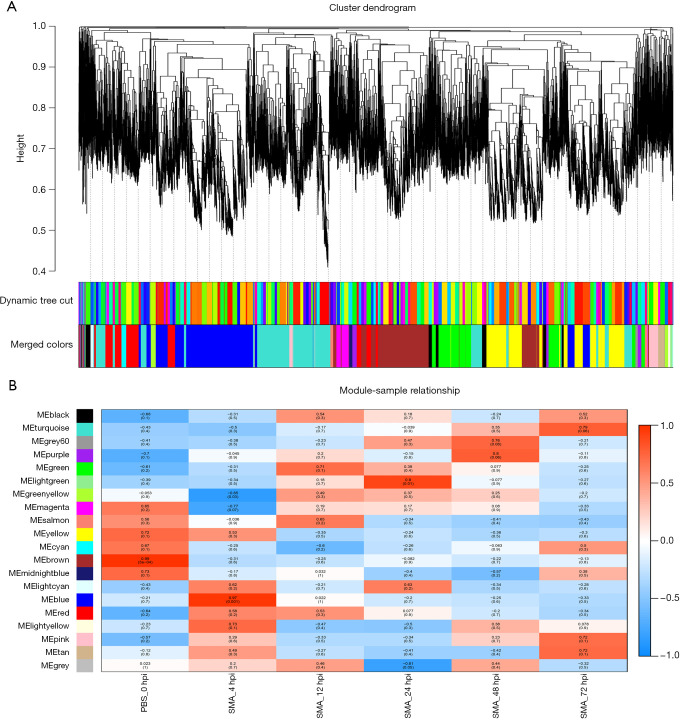
A mouse model of acute lethal SMA pneumonia was established in this study using aerosolized intratracheal inhalation, laying the groundwork for future SMA research. RNA-seq was then used to create a transcriptional profile of the lungs of the model mice at 0, 4, 12, 24, 48, and 72 hours post-infection (hpi). Mfuzz time clustering, weighted gene coexpression network analysis (WGCNA), and Immune Cell Abundance Identifier for mouse (ImmuCellAI-mouse) were used to analyze RNA-seq data.
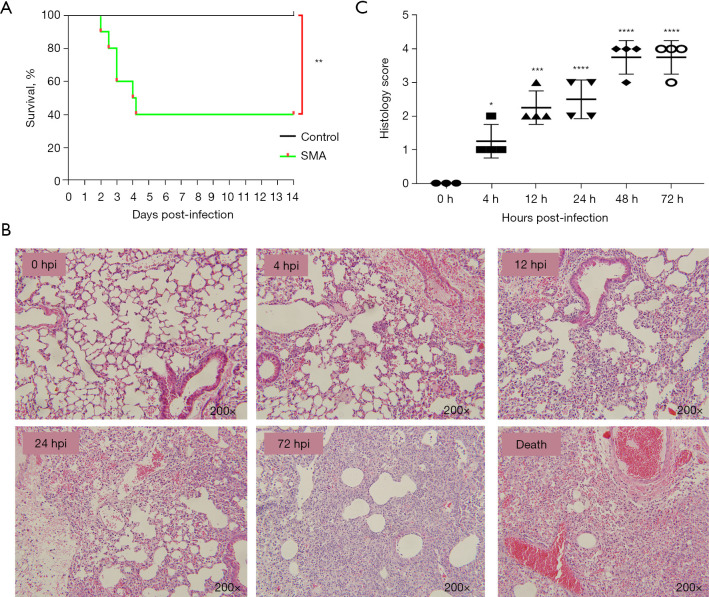
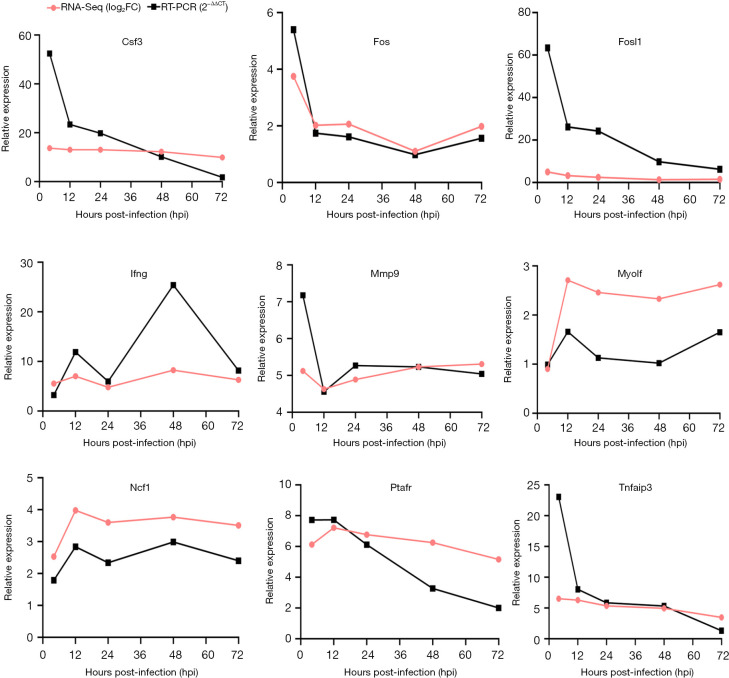
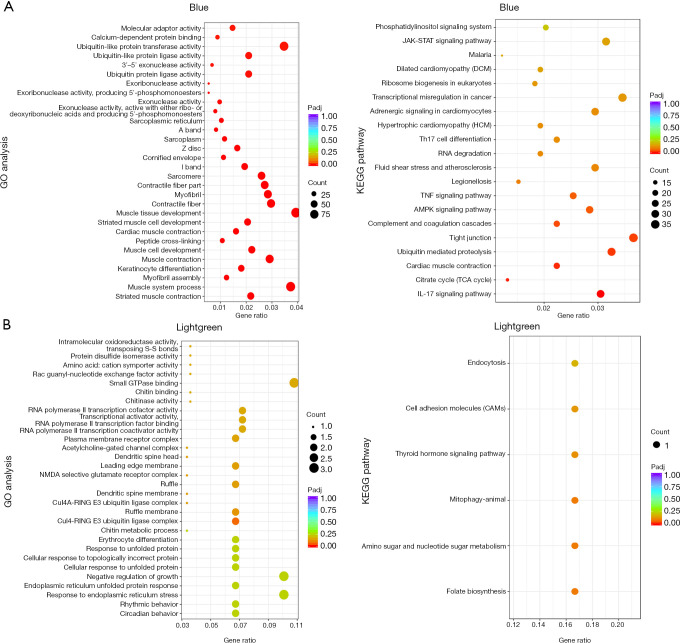
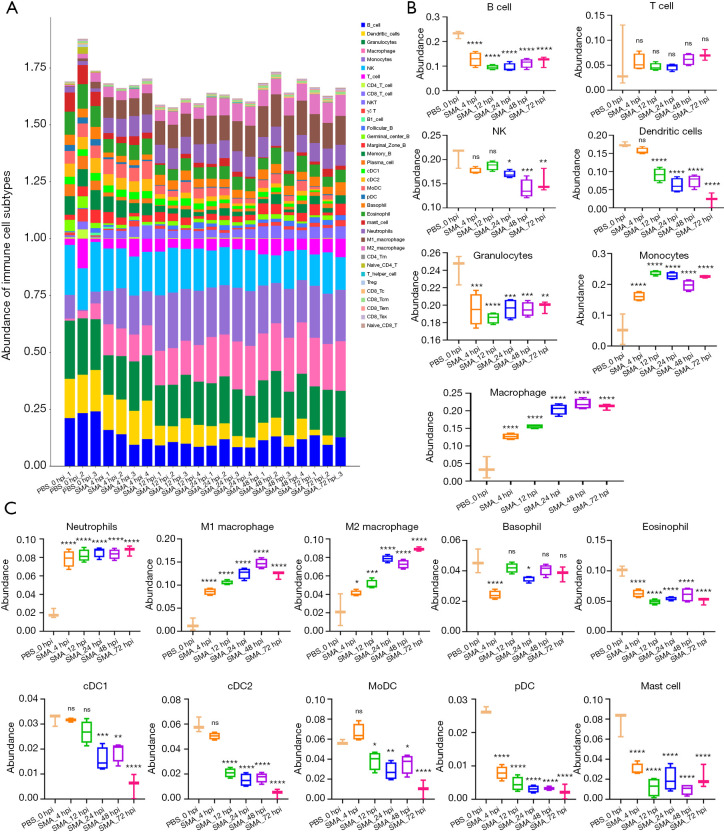
A gradual change in the lung transcriptional profile was observed, which was consistent with the expected disease progression. At 4 hpi, the expression of genes related to the acute phase inflammatory response increased, as predicted abundance of innate immune cells. At this stage, an increased demand for energy was also observed, including an increase in the expression of genes involved in circulation, muscle function and mitochondrial respiratory chain function. The expression of genes associated with endoplasmic reticulum stress (ERS) and autophagy increased at 24 hpi. Unlike the number of natural killer (NK) cells following most bacterial lung infections, the abundance of NK cells decreased following infection with SMA. The expression levels of , , , , , , , and 2 were high and previously unreported in SMA pneumonia, and they may be important targets for future studies.
To our knowledge, this is the first study to investigate the pulmonary transcriptional response to SMA infection. The findings shed light on the molecular mechanisms underlying the pathogenesis of SMA pneumonia, which may aid in the development of therapies to reduce the occurrence of SMA pulmonary infection.
嗜麦芽窄食单胞菌(SMA)已成为一种能够引起机会性感染和医院感染的重要病原体。我们通过气管内雾化吸入,对肺部感染SMA的小鼠肺组织随时间进行RNA测序(RNA-seq),以研究SMA感染肺组织中的转录谱变化。
本研究通过气管内雾化吸入建立了急性致死性SMA肺炎小鼠模型,为未来的SMA研究奠定了基础。然后使用RNA-seq创建感染后0、4、12、24、48和72小时(hpi)模型小鼠肺组织的转录谱。使用Mfuzz时间聚类、加权基因共表达网络分析(WGCNA)和小鼠免疫细胞丰度标识符(ImmuCellAI-小鼠)分析RNA-seq数据。
观察到肺转录谱的逐渐变化,这与预期的疾病进展一致。在感染后4小时,与急性期炎症反应相关的基因表达增加,如先天免疫细胞的预测丰度。在此阶段,还观察到能量需求增加,包括参与循环、肌肉功能和线粒体呼吸链功能的基因表达增加。与内质网应激(ERS)和自噬相关的基因表达在感染后24小时增加。与大多数细菌性肺部感染后自然杀伤(NK)细胞的数量不同,感染SMA后NK细胞的丰度下降。 、 、 、 、 、 、 、 和2的表达水平很高,且在SMA肺炎中以前未报道过,它们可能是未来研究的重要靶点。
据我们所知,这是第一项研究SMA感染肺部转录反应的研究。这些发现揭示了SMA肺炎发病机制的分子机制,这可能有助于开发减少SMA肺部感染发生的治疗方法。