Department of Computer Science and Engineering, University of California San Diego, La Jolla, CA, USA.
Department of Medicine, University of California San Diego, La Jolla, CA, USA.
Nat Commun. 2023 Oct 23;14(1):6711. doi: 10.1038/s41467-023-42278-3.
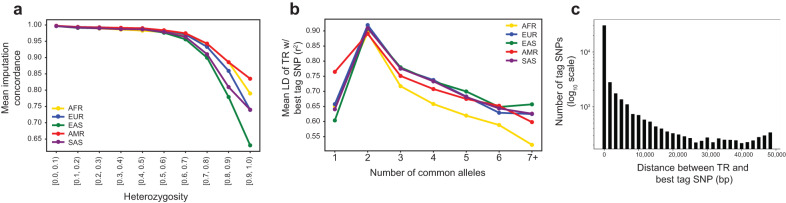
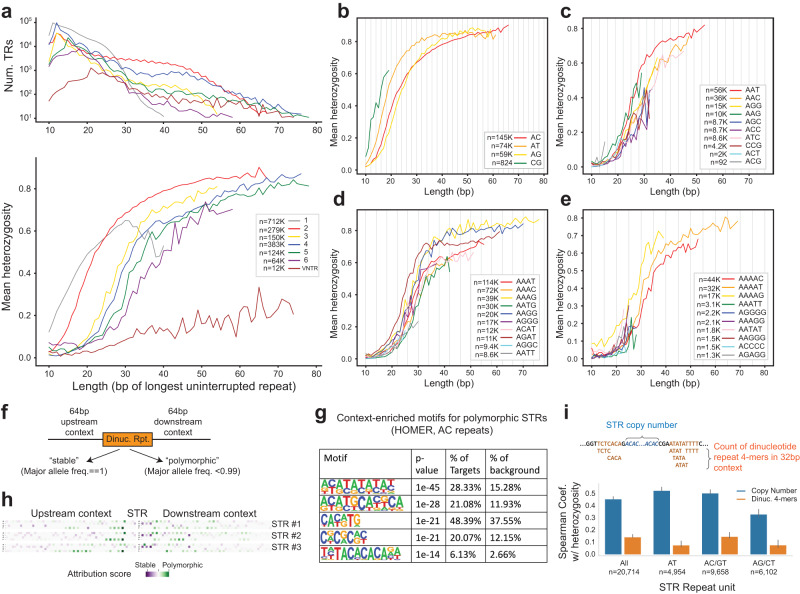
Tandem repeats (TRs) represent one of the largest sources of genetic variation in humans and are implicated in a range of phenotypes. Here we present a deep characterization of TR variation based on high coverage whole genome sequencing from 3550 diverse individuals from the 1000 Genomes Project and H3Africa cohorts. We develop a method, EnsembleTR, to integrate genotypes from four separate methods resulting in high-quality genotypes at more than 1.7 million TR loci. Our catalog reveals novel sequence features influencing TR heterozygosity, identifies population-specific trinucleotide expansions, and finds hundreds of novel eQTL signals. Finally, we generate a phased haplotype panel which can be used to impute most TRs from nearby single nucleotide polymorphisms (SNPs) with high accuracy. Overall, the TR genotypes and reference haplotype panel generated here will serve as valuable resources for future genome-wide and population-wide studies of TRs and their role in human phenotypes.
串联重复序列(TRs)是人类遗传变异的最大来源之一,与多种表型相关。本研究基于 1000 基因组计划和 H3Africa 队列中 3550 名个体的高覆盖全基因组测序,对 TR 变异进行了深入分析。我们开发了一种名为 EnsembleTR 的方法,整合了来自四种不同方法的基因型,从而在超过 170 万个 TR 位点获得了高质量的基因型。我们的目录揭示了影响 TR 杂合性的新序列特征,确定了人群特异性三核苷酸扩展,并发现了数百个新的 eQTL 信号。最后,我们生成了一个相位单倍型面板,可用于通过附近的单核苷酸多态性(SNPs)对大多数 TR 进行高精度推断。总体而言,这里生成的 TR 基因型和参考单倍型面板将成为未来 TR 及其在人类表型中作用的全基因组和全人群研究的宝贵资源。