Department of Anesthesiology, The First Hospital of Jilin University, No. 1 Xinmin St., Changchun, 130021, China.
Mol Neurobiol. 2024 Apr;61(4):2313-2335. doi: 10.1007/s12035-023-03695-z. Epub 2023 Oct 24.
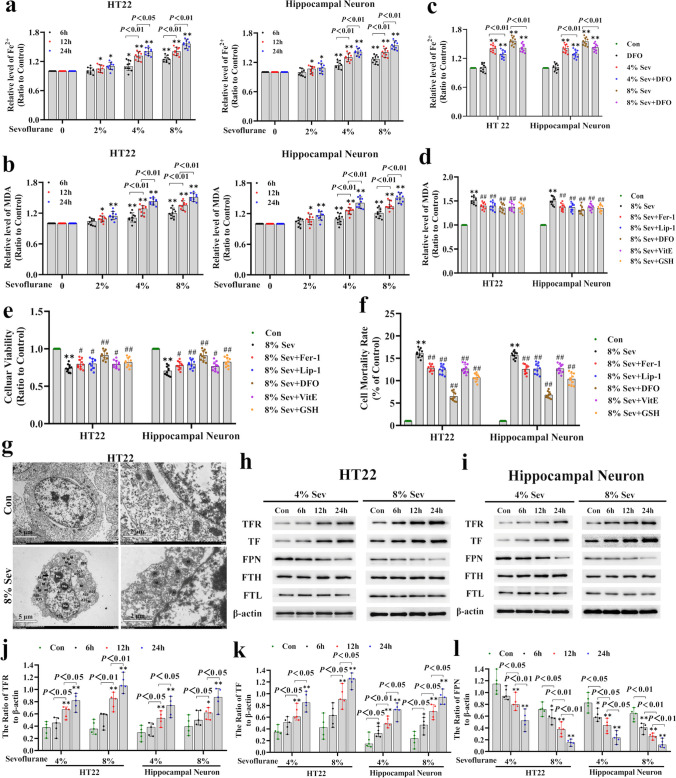
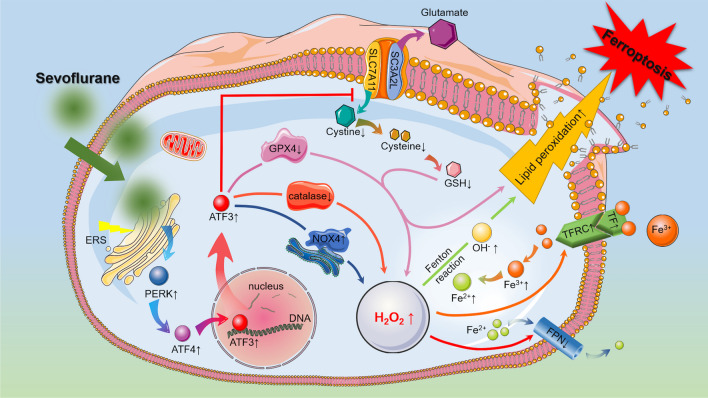
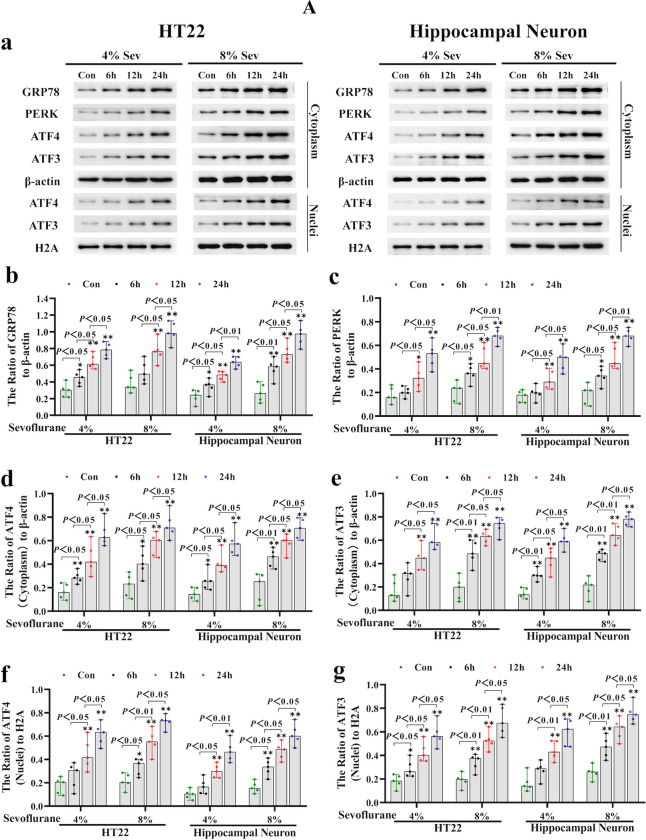
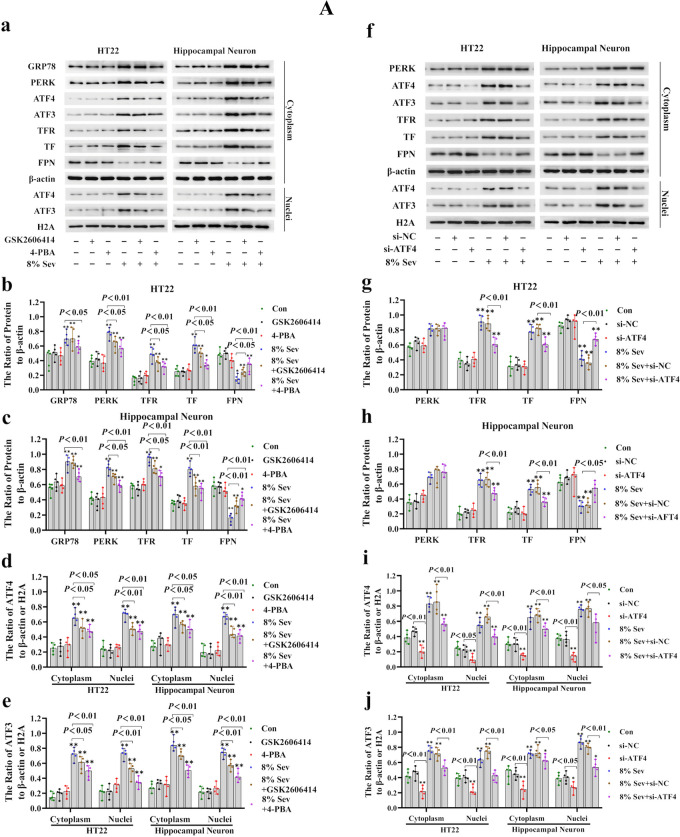
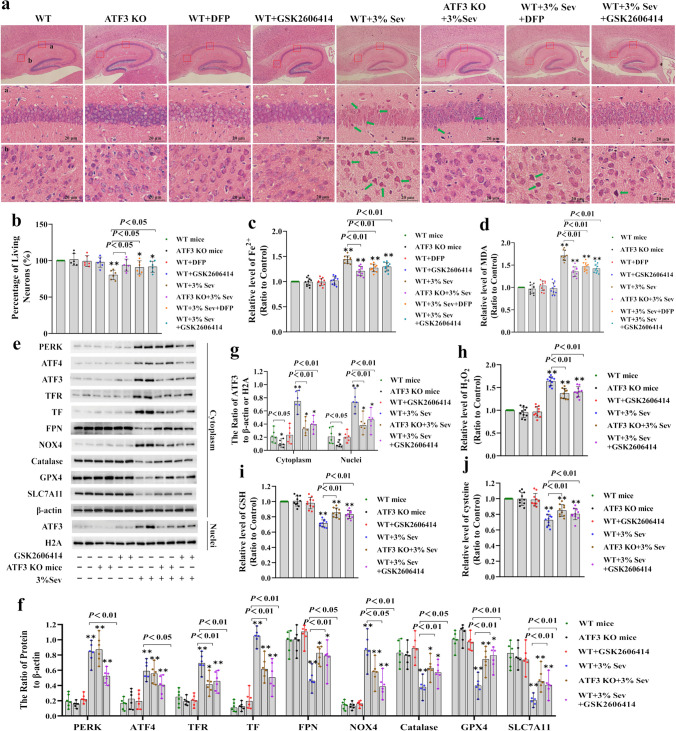
Neuronal cell death is acknowledged as the primary pathological basis underlying developmental neurotoxicity in response to sevoflurane exposure, but the exact mechanism remains unclear. Ferroptosis is a form of programmed cell death characterized by iron-dependent lipid peroxidation that is driven by hydrogen peroxide (HO) and ferrous iron through the Fenton reaction and participates in the pathogenesis of multiple neurological diseases. As stress response factor, activating transcription factor 3 (ATF3) can be activated by the PERK/ATF4 pathway during endoplasmic reticulum (ER) stress, followed by increased intracellular HO, which is involved in regulation of apoptosis, autophagy, and ferroptosis. Here, we investigated whether ferroptosis and ATF3 activation were implicated in sevoflurane-induced neuronal cell death in the developing brain. The results showed that sevoflurane exposure induced neuronal death as a result of iron-dependent lipid peroxidation damage secondary to HO accumulation and ferrous iron increase, which was consistent with the criteria for ferroptosis. Furthermore, we observed that increases in iron and HO induced by sevoflurane exposure were associated with the upregulation and nuclear translocation of ATF3 in response to ER stress. Knockdown of ATF3 expression alleviated iron-dependent lipid peroxidation, which prevented sevoflurane-induced neuronal ferroptosis. Mechanistically, ATF3 promoted sevoflurane-induced HO accumulation by activating NOX4 and suppressing catalase, GPX4, and SLC7A11 expression. Additionally, an increase in HO was accompanied by the upregulation of TFR and TF and downregulation of FPN, which linked iron overload to ferroptosis induced by sevoflurane. Taken together, our results demonstrated that ER stress-mediated ATF3 activation contributed to sevoflurane-induced neuronal ferroptosis via HO accumulation and the resultant iron overload.
神经元细胞死亡被认为是七氟醚暴露导致发育性神经毒性的主要病理基础,但确切机制尚不清楚。铁死亡是一种程序性细胞死亡形式,其特征是依赖铁的脂质过氧化,由过氧化氢 (HO) 和二价铁通过芬顿反应驱动,并参与多种神经疾病的发病机制。作为应激反应因子,激活转录因子 3 (ATF3) 可以在内质网 (ER) 应激期间被 PERK/ATF4 途径激活,随后细胞内 HO 增加,参与凋亡、自噬和铁死亡的调节。在这里,我们研究了铁死亡和 ATF3 激活是否与七氟醚诱导的发育期大脑神经元细胞死亡有关。结果表明,七氟醚暴露诱导神经元死亡是由于 HO 积累和二价铁增加导致的铁依赖性脂质过氧化损伤,这与铁死亡的标准一致。此外,我们观察到七氟醚暴露引起的铁和 HO 的增加与 ER 应激时 ATF3 的上调和核转位有关。ATF3 表达的敲低减轻了铁依赖性脂质过氧化,从而防止了七氟醚诱导的神经元铁死亡。在机制上,ATF3 通过激活 NOX4 和抑制过氧化氢酶、GPX4 和 SLC7A11 的表达来促进七氟醚诱导的 HO 积累。此外,HO 的增加伴随着 TFR 和 TF 的上调和 FPN 的下调,这将铁过载与七氟醚诱导的铁死亡联系起来。总之,我们的结果表明,ER 应激介导的 ATF3 激活通过 HO 积累和由此产生的铁过载导致七氟醚诱导的神经元铁死亡。