Giraldo Jorge Navarro, Hrubý Jakub, Vavrečková Šárka, Fellner Ondřej F, Havlíček Lubomír, Henry DaVonne, de Silva Shehan, Herchel Radovan, Bartoš Miroslav, Šalitroš Ivan, Santana Vinicius T, Barbara Paola, Nemec Ivan, Neugebauer Petr
Central European Institute of Technology, CEITEC BUT, Purkyňova 656/123, 61200 Brno, Czech Republic.
Institute of Physical Engineering, Faculty of Mechanical Engineering, Brno University of Technology, Technická 2, 61669 Brno, Czech Republic.
Phys Chem Chem Phys. 2023 Nov 8;25(43):29516-29530. doi: 10.1039/d3cp01426f.
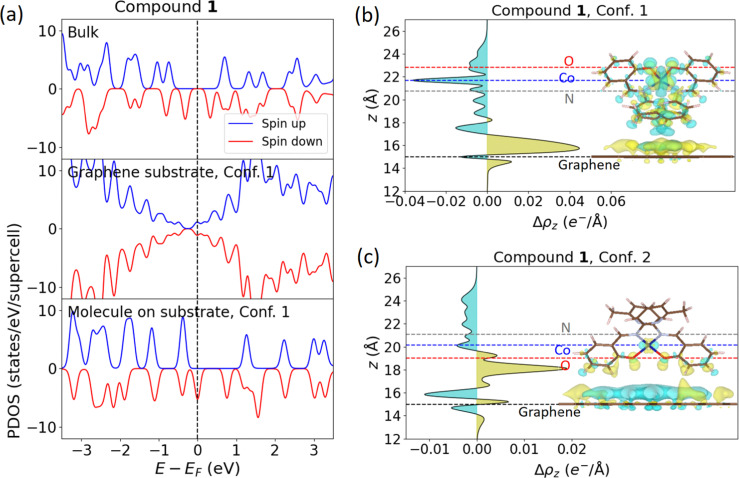
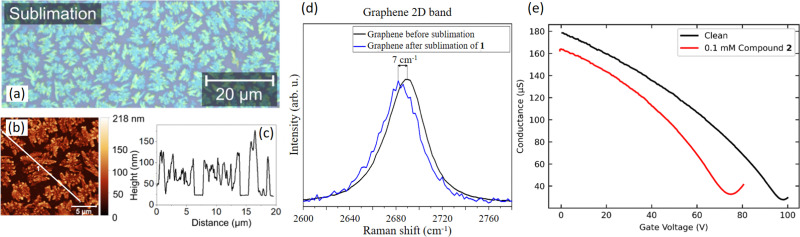
We present a theoretical and experimental study of two tetracoordinate Co(II)-based complexes with semi-coordination interactions, , non-covalent interactions involving the central atom. We argue that such interactions enhance the thermal and structural stability of the compounds, making them appropriate for deposition on substrates, as demonstrated by their successful deposition on graphene. DC magnetometry and high-frequency electron spin resonance (HF-ESR) experiments revealed an axial magnetic anisotropy and weak intermolecular antiferromagnetic coupling in both compounds, supported by theoretical predictions from complete active space self-consistent field calculations complemented by N-electron valence state second-order perturbation theory (CASSCF-NEVPT2), and broken-symmetry density functional theory (BS-DFT). AC magnetometry demonstrated that the compounds are field-induced single-ion magnets (SIMs) at applied static magnetic fields, with slow relaxation of magnetization governed by a combination of quantum tunneling, Orbach, and direct relaxation mechanisms. The structural stability under ambient conditions and after deposition was confirmed by X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy. Theoretical modeling by DFT of different configurations of these systems on graphene revealed n-type doping of graphene originating from electron transfer from the deposited molecules, confirmed by electrical transport measurements and Raman spectroscopy.
我们展示了对两种具有半配位相互作用的四配位钴(II)基配合物的理论和实验研究,这种半配位相互作用是涉及中心原子的非共价相互作用。我们认为,这种相互作用增强了化合物的热稳定性和结构稳定性,使其适合沉积在基底上,在石墨烯上的成功沉积就证明了这一点。直流磁强计和高频电子自旋共振(HF - ESR)实验揭示了两种化合物中均存在轴向磁各向异性和弱分子间反铁磁耦合,这得到了完全活性空间自洽场计算辅以N电子价态二阶微扰理论(CASSCF - NEVPT2)以及破缺对称性密度泛函理论(BS - DFT)的理论预测的支持。交流磁强计表明,在施加静态磁场时,这些化合物是场致单离子磁体(SIMs),其磁化强度的缓慢弛豫由量子隧穿、奥巴赫和直接弛豫机制共同控制。通过X射线光电子能谱(XPS)和拉曼光谱证实了在环境条件下以及沉积后的结构稳定性。通过密度泛函理论(DFT)对这些系统在石墨烯上的不同构型进行理论建模,揭示了源自沉积分子电子转移的石墨烯n型掺杂,这一点通过电输运测量和拉曼光谱得到了证实。