Shi Benjamin X, Zen Andrea, Kapil Venkat, Nagy Péter R, Grüneis Andreas, Michaelides Angelos
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, U.K.
Dipartimento di Fisica Ettore Pancini, Università di Napoli Federico II, Monte S. Angelo, I-80126 Napoli, Italy.
J Am Chem Soc. 2023 Nov 22;145(46):25372-25381. doi: 10.1021/jacs.3c09616. Epub 2023 Nov 10.
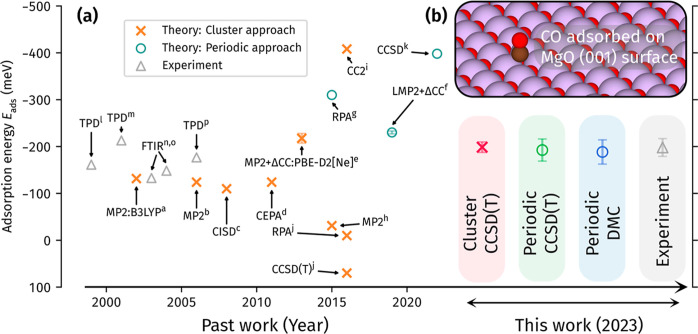
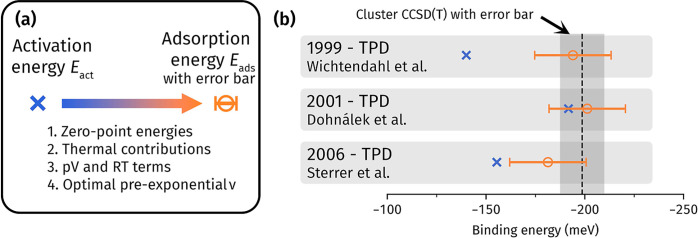
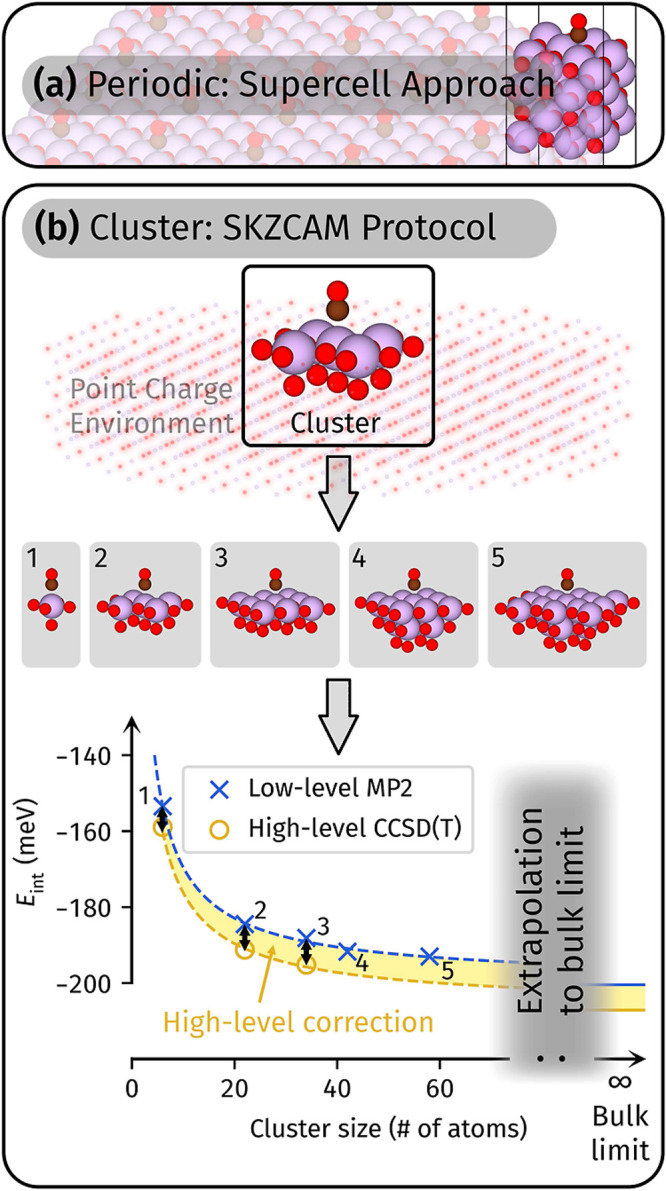
The adsorption energy of a molecule onto the surface of a material underpins a wide array of applications, spanning heterogeneous catalysis, gas storage, and many more. It is the key quantity where experimental measurements and theoretical calculations meet, with agreement being necessary for reliable predictions of chemical reaction rates and mechanisms. The prototypical molecule-surface system is CO adsorbed on MgO, but despite intense scrutiny from theory and experiment, there is still no consensus on its adsorption energy. In particular, the large cost of accurate many-body methods makes reaching converged theoretical estimates difficult, generating a wide range of values. In this work, we address this challenge, leveraging the latest advances in diffusion Monte Carlo (DMC) and coupled cluster with single, double, and perturbative triple excitations [CCSD(T)] to obtain accurate predictions for CO on MgO. These reliable theoretical estimates allow us to evaluate the inconsistencies in published temperature-programed desorption experiments, revealing that they arise from variations in employed pre-exponential factors. Utilizing this insight, we derive new experimental estimates of the (electronic) adsorption energy with a (more) precise pre-exponential factor. As a culmination of all of this effort, we are able to reach a consensus between multiple theoretical calculations and multiple experiments for the first time. In addition, we show that our recently developed cluster-based CCSD(T) approach provides a low-cost route toward achieving accurate adsorption energies. This sets the stage for affordable and reliable theoretical predictions of chemical reactions on surfaces to guide the realization of new catalysts and gas storage materials.
分子在材料表面的吸附能支撑着广泛的应用,涵盖多相催化、气体存储等等。它是实验测量与理论计算交汇的关键量,对于可靠预测化学反应速率和机理而言,两者达成一致是必要的。典型的分子 - 表面体系是一氧化碳吸附在氧化镁上,然而尽管理论和实验都进行了深入研究,但对于其吸附能仍未达成共识。特别是,精确的多体方法成本高昂,使得获得收敛的理论估计变得困难,从而产生了广泛的数值范围。在这项工作中,我们应对这一挑战,利用扩散蒙特卡罗(DMC)以及含单、双和微扰三重激发的耦合簇方法[CCSD(T)]的最新进展,来获得一氧化碳在氧化镁上的精确预测。这些可靠的理论估计使我们能够评估已发表的程序升温脱附实验中的不一致性,揭示出它们源于所采用的指前因子的变化。利用这一见解,我们用(更)精确的指前因子得出了(电子)吸附能的新实验估计值。经过所有这些努力,我们首次能够在多个理论计算和多个实验之间达成共识。此外,我们表明,我们最近开发的基于簇的CCSD(T)方法为获得精确的吸附能提供了一条低成本途径。这为表面化学反应的经济且可靠的理论预测奠定了基础,以指导新型催化剂和气体存储材料的研发。