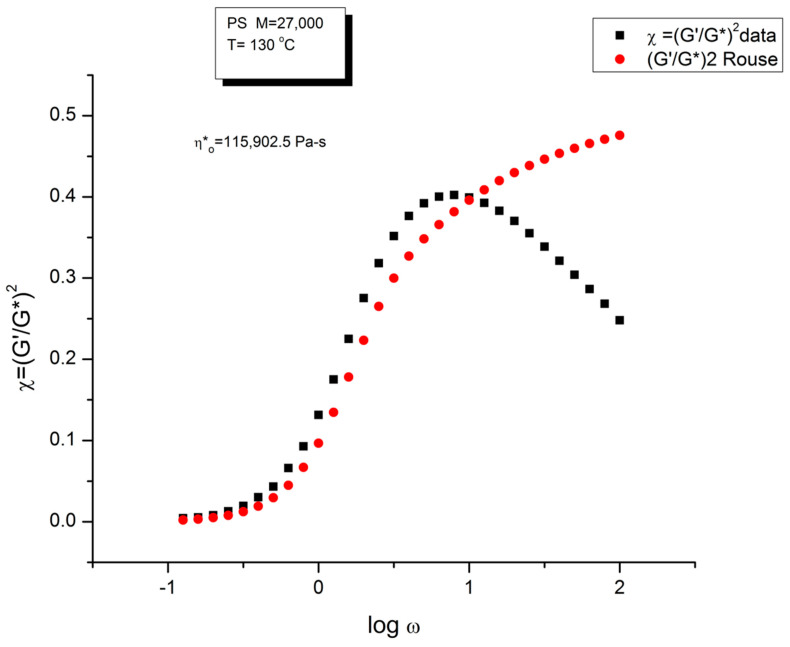
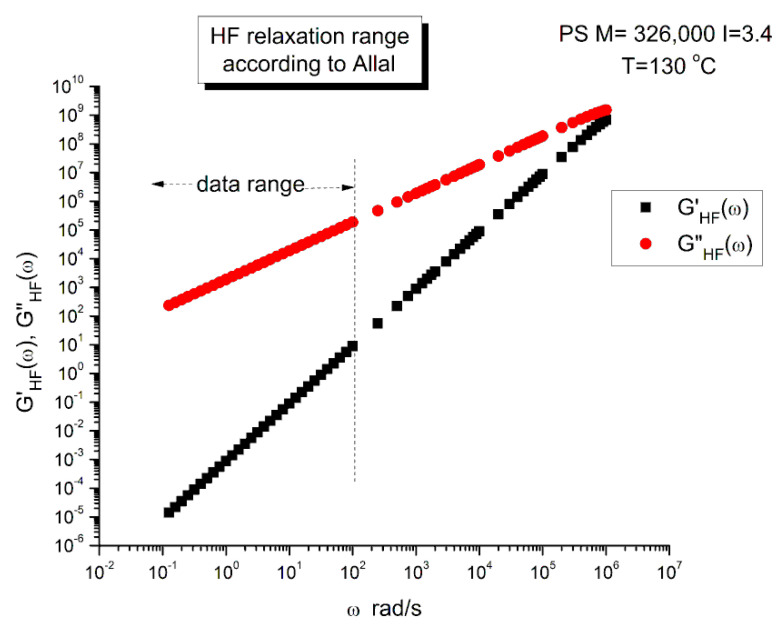
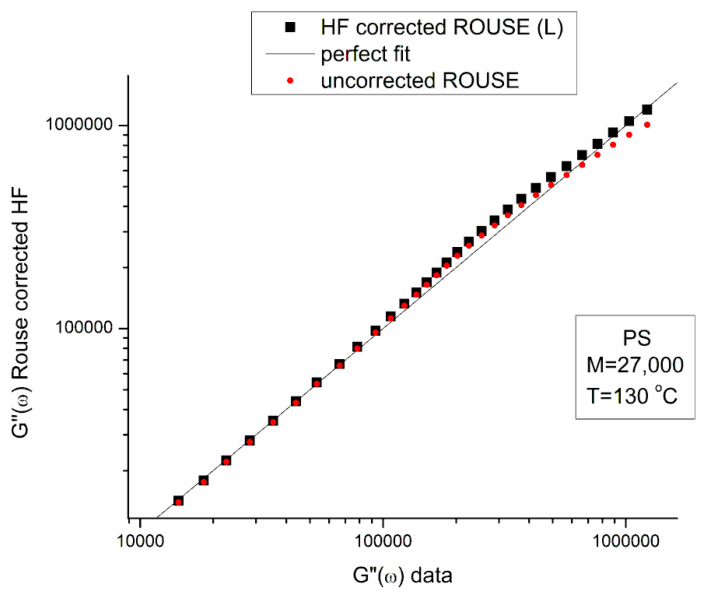
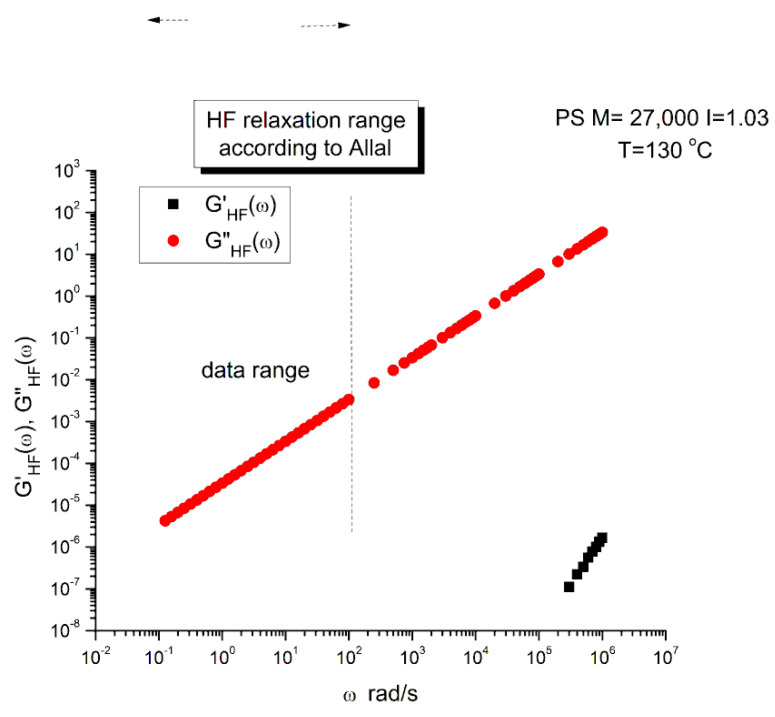
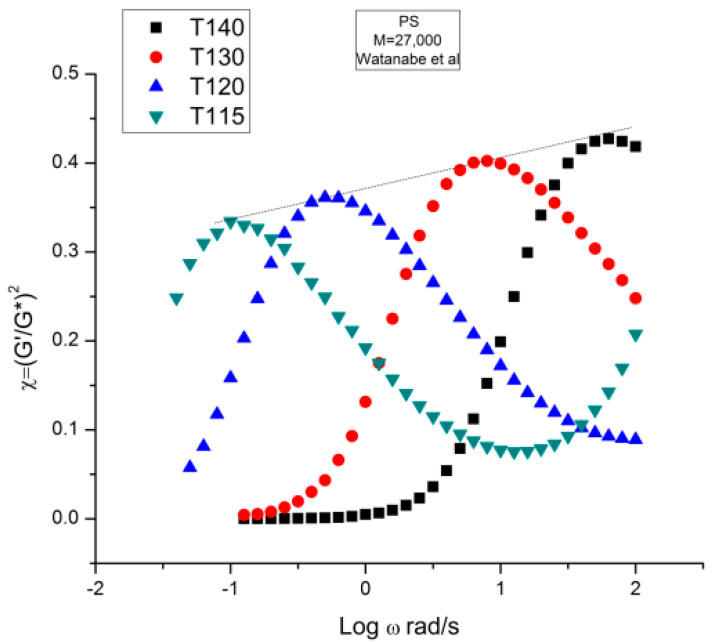
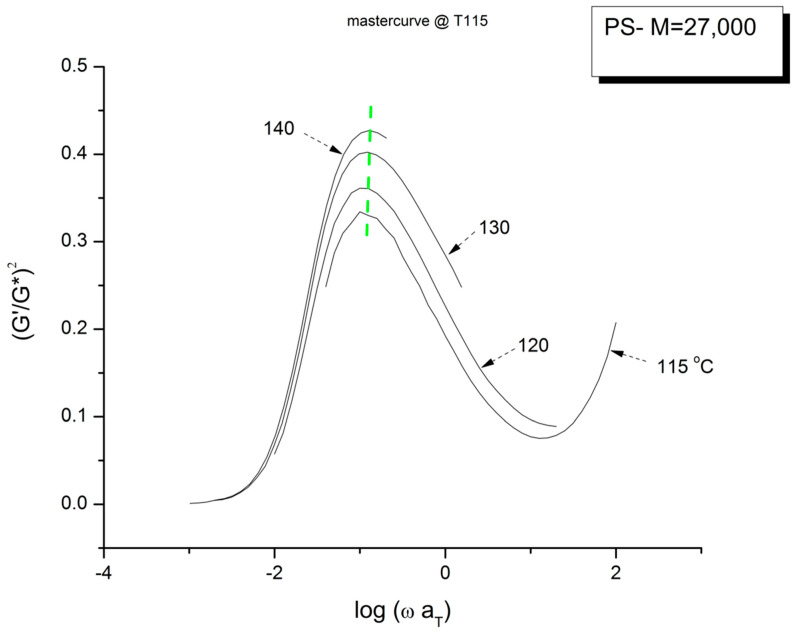
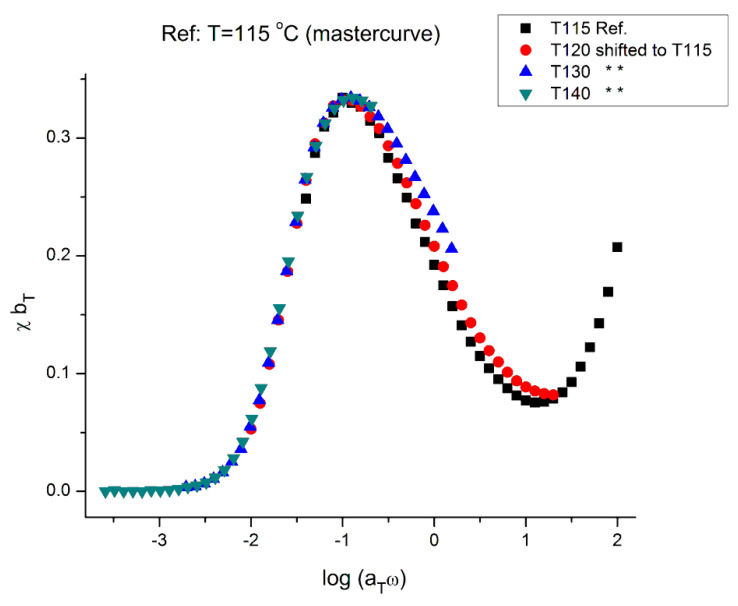
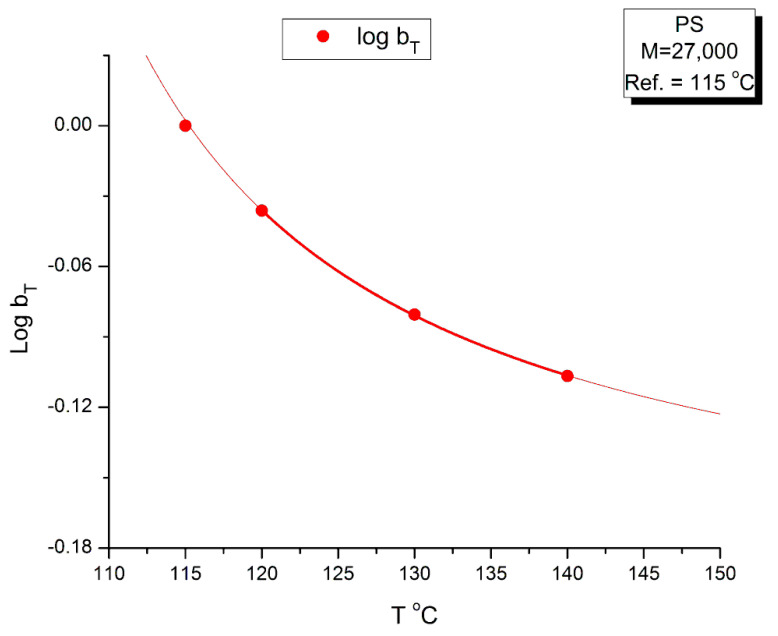
Ibar Jean Pierre
Polymat Institute, University of the Basque Country (UPV/EHU), 48013 Donostia-San Sebastian, Euskadi, Spain.
Polymers (Basel). 2023 Nov 2;15(21):4309. doi: 10.3390/polym15214309.
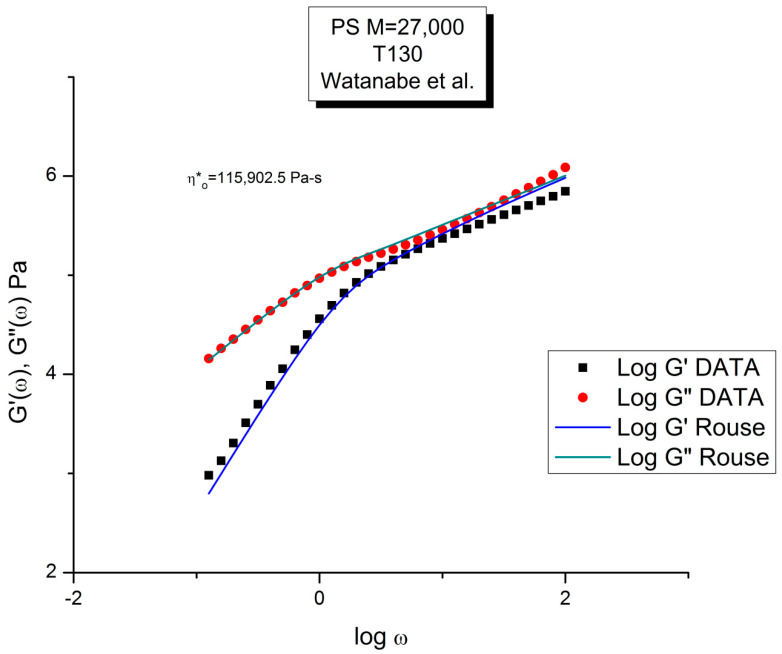
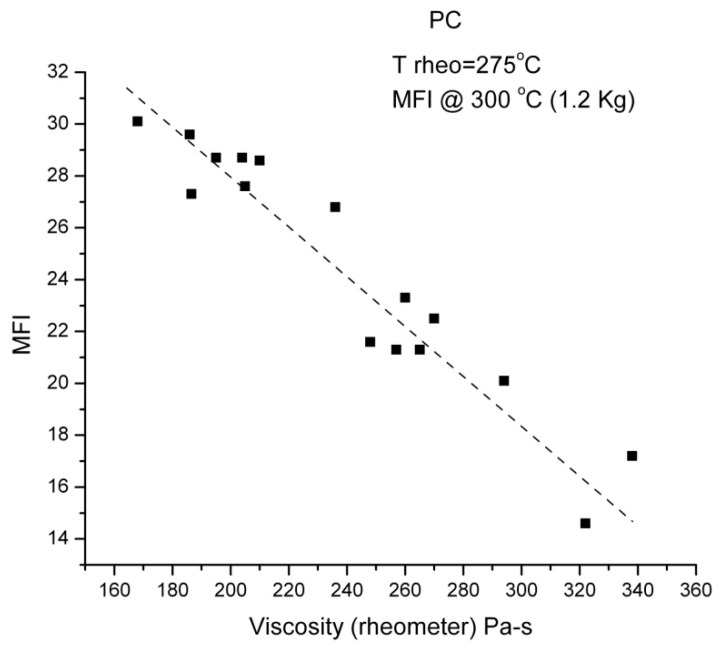
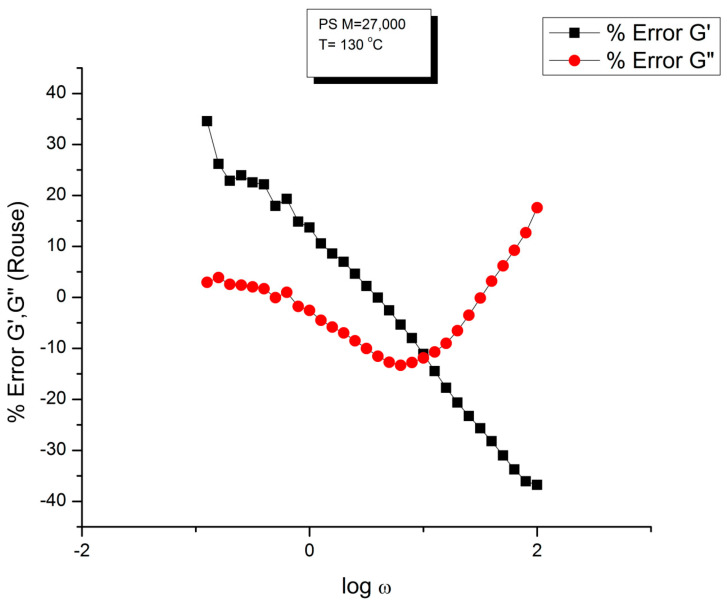
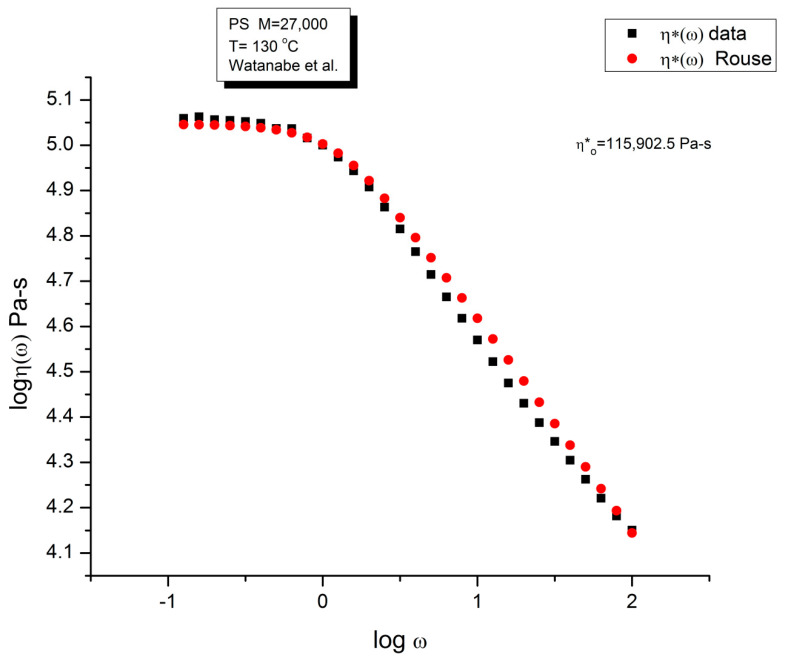
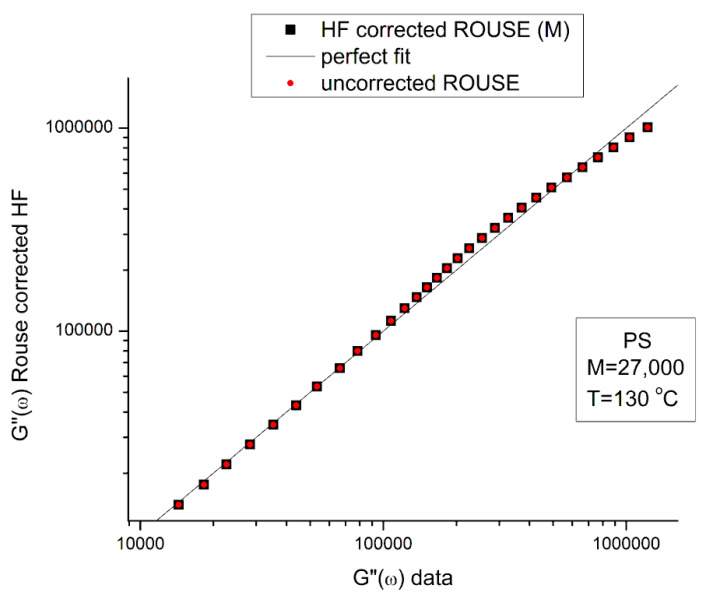
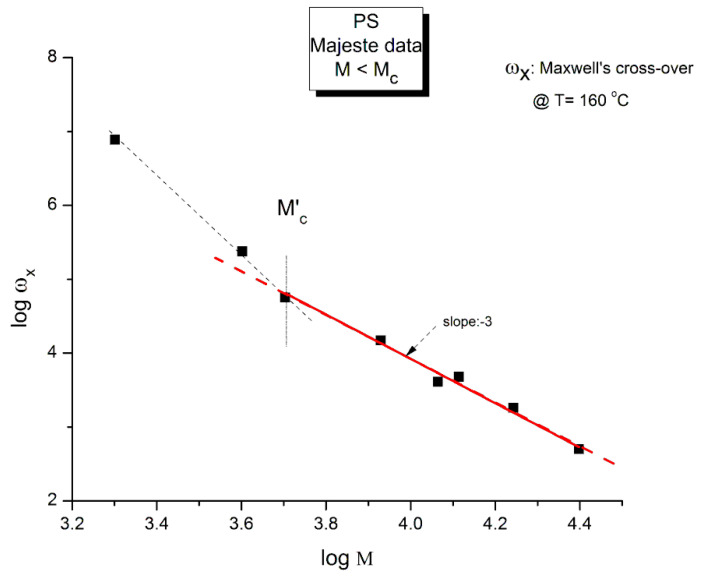
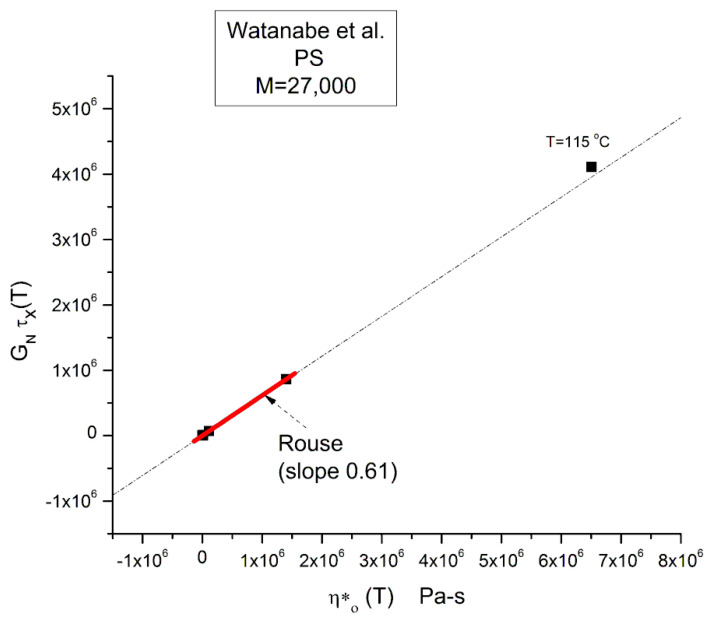
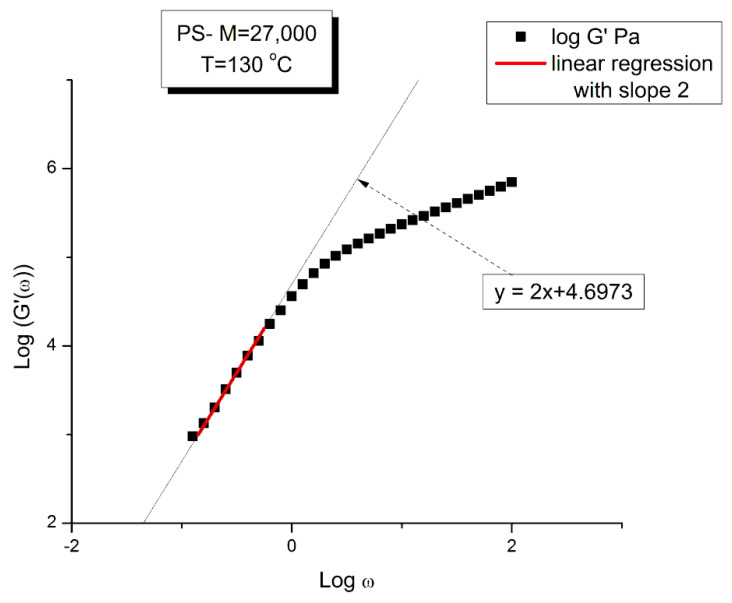
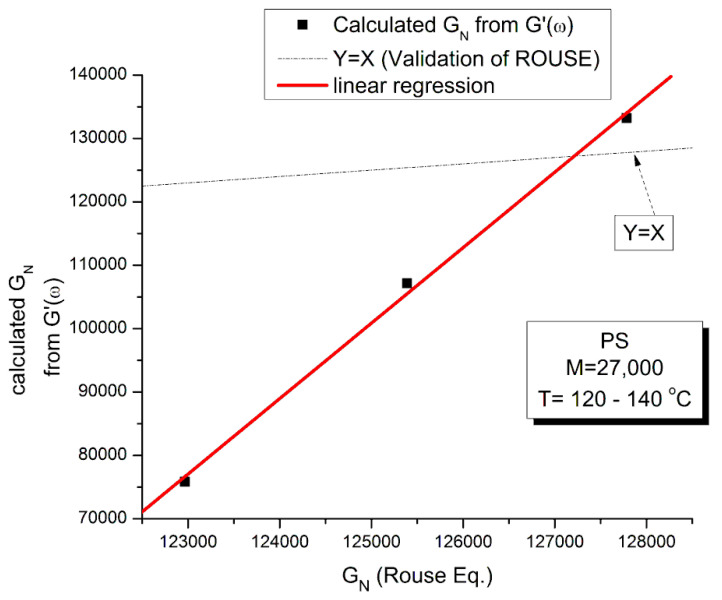
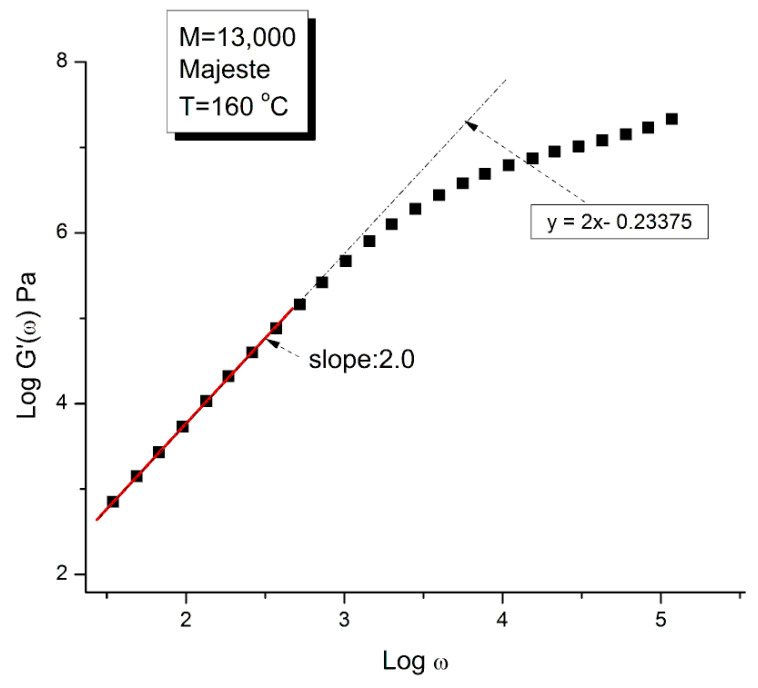
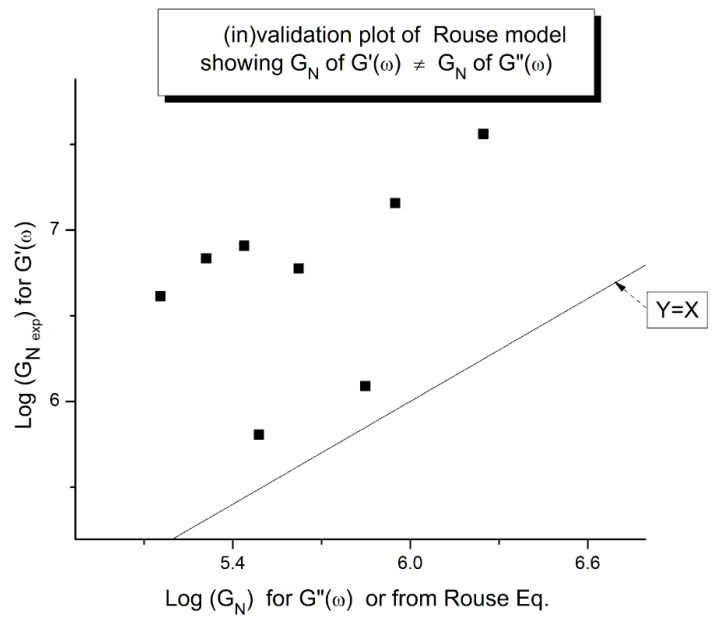
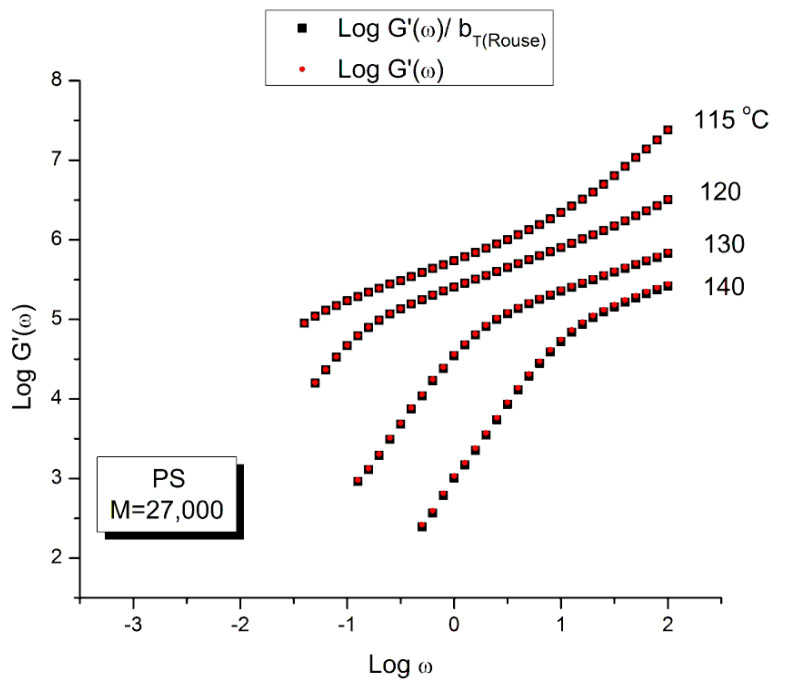
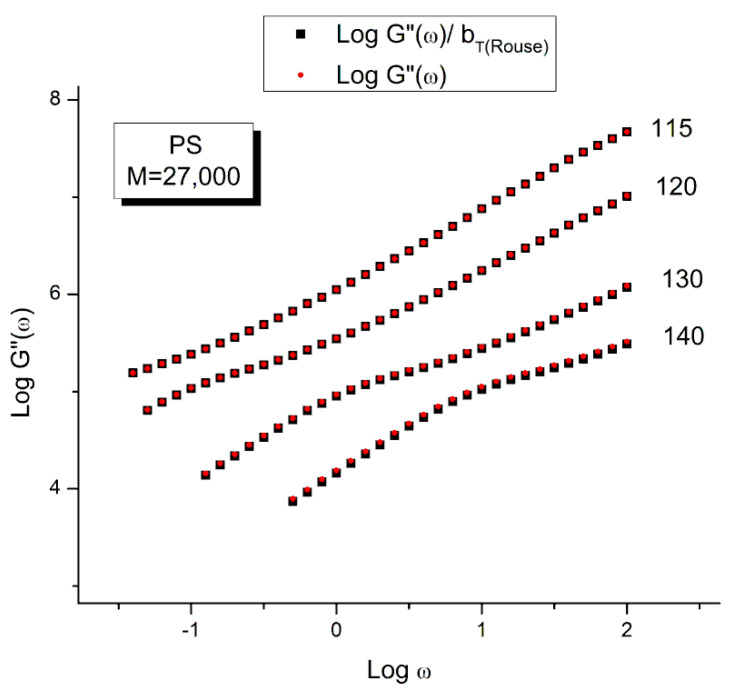
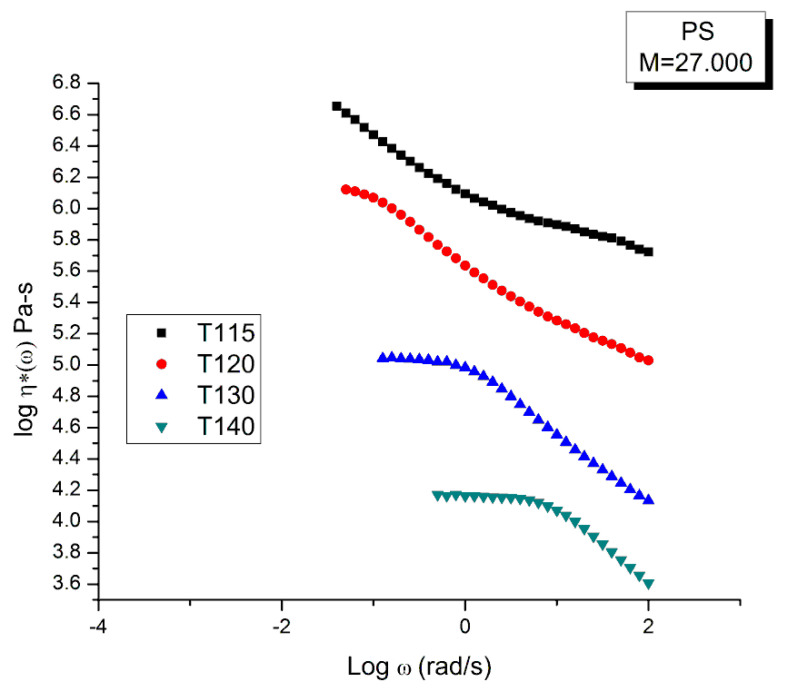
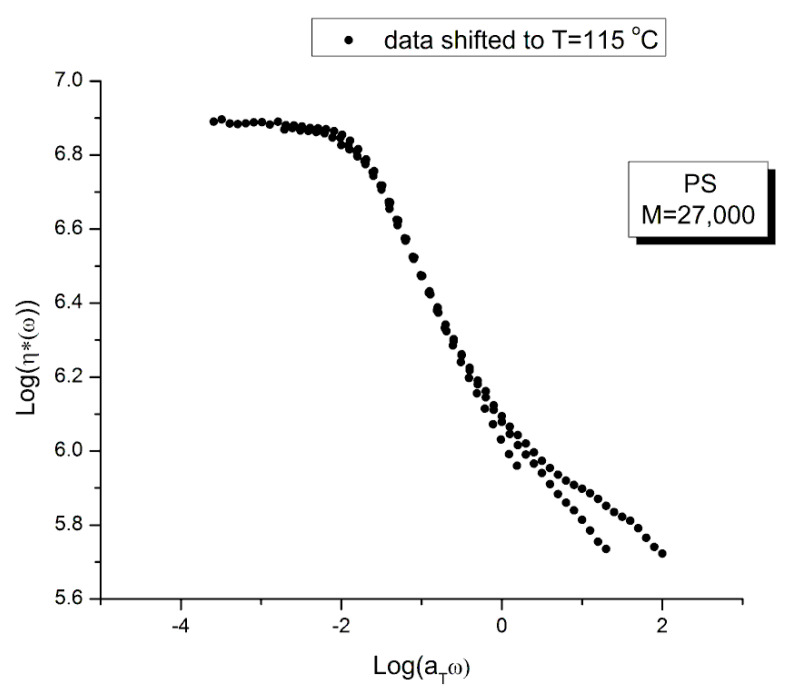
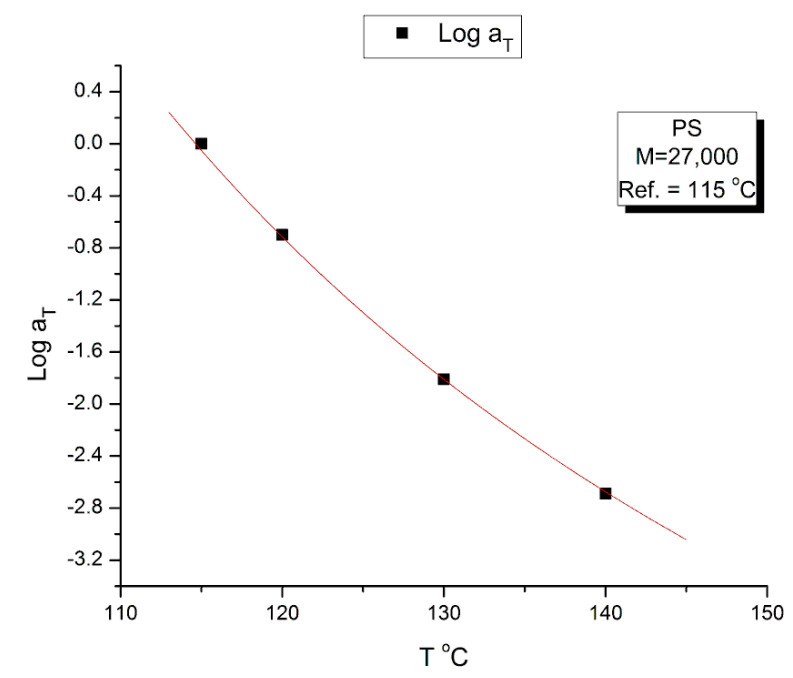
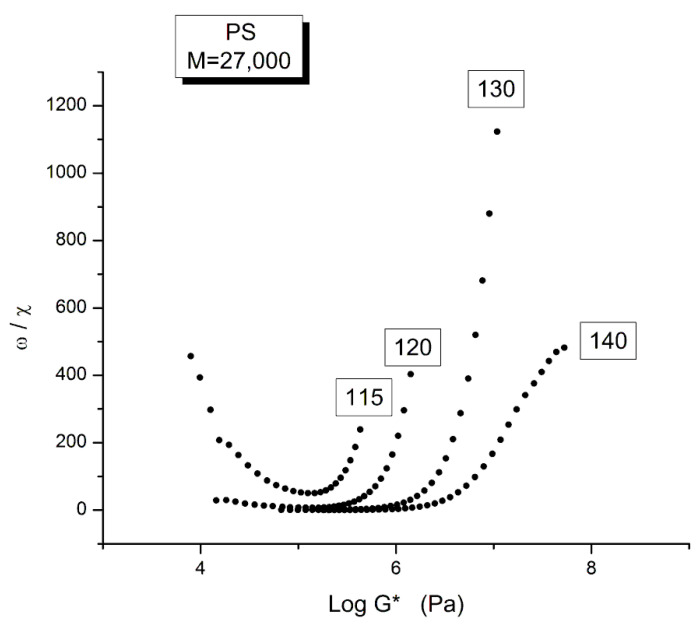
Staudinger taught us that macromolecules were made up of covalently bonded monomer repeat units chaining up as polymer chains. This paradigm is not challenged in this paper. The main question raised in polymer physics remains: how do these long chains interact and move as a group when submitted to shear deformation at high temperature when they are viscous liquids? The current consensus is that we need to distinguish two cases: the deformation of "un-entangled chains" for macromolecules with molecular weight, M, smaller than M, "the entanglement molecular weight", and the deformation of "entangled" chains for M > M. The current paradigm stipulates that the properties of polymers derive from the statistical characteristics of the macromolecule itself, the designated statistical system that defines the thermodynamic state of the polymer. The current paradigm claims that the viscoelasticity of un-entangled melts is well described by the Rouse model and that the entanglement issues raised when M > Me, are well understood by the reptation model introduced by de Gennes and colleagues. Both models can be classified in the category of "chain dynamics statistics". In this paper, we examine in detail the failures and the current challenges facing the current paradigm of polymer rheology: the Rouse model for un-entangled melts, the reptation model for entangled melts, the time-temperature superposition principle, the strain-induced time dependence of viscosity, shear-refinement and sustained-orientation. The basic failure of the current paradigm and its inherent inability to fully describe the experimental reality is documented in this paper. In the discussion and conclusion sections of the paper, we suggest that a different solution to explain the viscoelasticity of polymer chains and of their "entanglement" is needed. This requires a change in paradigm to describe the dynamics of the interactions within the chains and across the chains. A brief description of our currently proposed open dissipative statistical approach, "the Grain-Field Statistics", is presented.
施陶丁格告诉我们,大分子是由通过共价键连接的单体重复单元组成的,这些单元像聚合物链一样连接在一起。本文并未对这一范式提出挑战。聚合物物理学中提出的主要问题仍然是:当这些长链在高温下作为粘性液体受到剪切变形时,它们如何作为一个整体相互作用和移动?目前的共识是,我们需要区分两种情况:对于分子量M小于“缠结分子量”Me的大分子,“未缠结链”的变形;以及对于M > Me的“缠结”链的变形。当前的范式规定,聚合物的性质源于大分子本身的统计特性,即定义聚合物热力学状态的指定统计系统。当前的范式认为,未缠结熔体的粘弹性可以通过劳斯模型很好地描述,而当M > Me时出现的缠结问题可以通过德热纳及其同事提出的爬行模型很好地理解。这两个模型都可以归类为“链动力学统计”范畴。在本文中,我们详细研究了当前聚合物流变学范式所面临的失败和当前挑战:未缠结熔体的劳斯模型、缠结熔体的爬行模型、时间 - 温度叠加原理、应变诱导的粘度时间依赖性、剪切细化和持续取向。本文记录了当前范式的基本失败及其无法完全描述实验现实的内在缺陷。在本文的讨论和结论部分,我们建议需要一种不同的解决方案来解释聚合物链及其“缠结”的粘弹性。这需要改变范式来描述链内和链间相互作用的动力学。本文简要介绍了我们目前提出的开放耗散统计方法,即“粒场统计”。