Guttenberg M A, Vose A T, Birukova A, Lewars K, Cumming R I, Albright M C, Mark J I, Salazar C J, Swaminathan S, Yu Z, Sokolenko Yu V, Bunyan E, Yaeger M J, Fessler M B, Que L G, Gowdy K M, Misharin A V, Tighe R M
Division of Pulmonary, Allergy and Critical Care Medicine, Department of Medicine, Duke University, Durham, NC.
Department of Health Sciences, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA.
bioRxiv. 2023 Nov 6:2023.11.06.565865. doi: 10.1101/2023.11.06.565865.
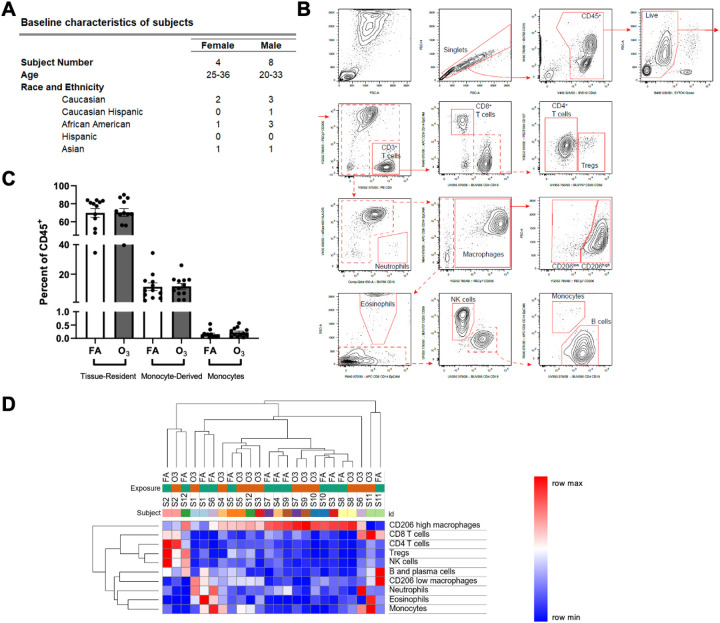
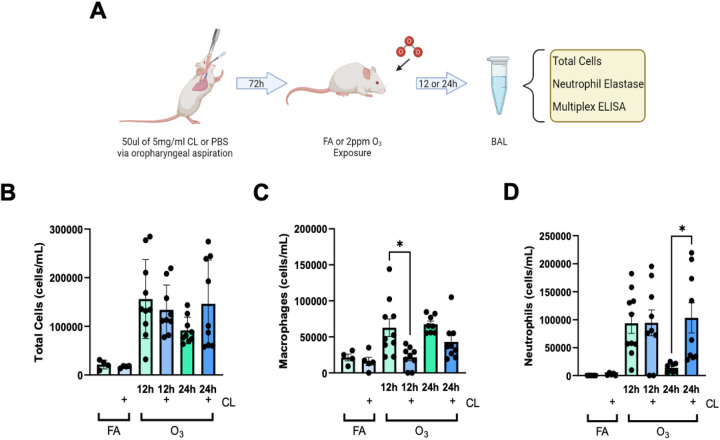
Lung inflammation, caused by acute exposure to ozone (O) - one of the six criteria air pollutants - is a significant source of morbidity in susceptible individuals. Alveolar macrophages (AMØs) are the most abundant immune cells in the normal lung and their number increases following O exposure. However, the role of AMØs in promoting or limiting O-induced lung inflammation has not been clearly defined. Here, we used a mouse model of acute O exposure, lineage tracing, genetic knockouts, and data from O-exposed human volunteers to define the role and ontogeny of AMØs during acute O exposure. Lineage tracing experiments showed that 12, 24, and 72 h after exposure to O (2 ppm) for 3h all AMØs were tissue-resident origin. Similarly, in humans exposed to FA and O (200 ppb) for 135 minutes, we did not observe ~21h post-exposure an increase in monocyte-derived AMØs by flow cytometry. Highlighting a role for tissue-resident AMØs, we demonstrate that depletion of tissue-resident AMØs with clodronate-loaded liposomes led to persistence of neutrophils in the alveolar space after O exposure, suggesting that impaired neutrophil clearance (i.e., efferocytosis) leads to prolonged lung inflammation. Moreover, depletion of tissue-resident AMØ demonstrated reduced clearance of intratracheally instilled apoptotic Jurkat cells, consistent with reduced efferocytosis. Genetic ablation of MerTK - a key receptor involved in efferocytosis - also resulted in impaired clearance of apoptotic neutrophils followed O exposure. Overall, these findings underscore the pivotal role of tissue-resident AMØs in resolving O-induced inflammation via MerTK-mediated efferocytosis.
急性暴露于臭氧(O₃,六种标准空气污染物之一)所引起的肺部炎症,是易感个体发病的一个重要原因。肺泡巨噬细胞(AMØs)是正常肺组织中数量最多的免疫细胞,臭氧暴露后其数量会增加。然而,AMØs在促进或限制臭氧诱导的肺部炎症中所起的作用尚未明确界定。在此,我们利用急性臭氧暴露小鼠模型、谱系追踪、基因敲除以及臭氧暴露人类志愿者的数据,来明确急性臭氧暴露期间AMØs的作用和起源。谱系追踪实验表明,在暴露于3 ppm臭氧3小时后的12、24和72小时,所有AMØs均来源于组织驻留细胞。同样,在暴露于200 ppb甲醛和臭氧135分钟的人类志愿者中,暴露后约21小时,我们通过流式细胞术未观察到单核细胞来源的AMØs增加。通过用负载氯膦酸盐的脂质体耗尽组织驻留AMØs,我们证明了其在臭氧暴露后肺泡空间中嗜中性粒细胞的持续存在,这突出了组织驻留AMØs的作用,表明嗜中性粒细胞清除功能受损(即吞噬作用)会导致肺部炎症延长。此外,耗尽组织驻留AMØs表明气管内注入的凋亡Jurkat细胞的清除减少,这与吞噬作用降低一致。MerTK(参与吞噬作用的关键受体)的基因敲除也导致臭氧暴露后凋亡嗜中性粒细胞的清除受损。总体而言,这些发现强调了组织驻留AMØs在通过MerTK介导的吞噬作用解决臭氧诱导的炎症中的关键作用。