Department of Computer Science, Florida State University, Tallahassee, Florida 32306, USA.
Department of Biological Science, Florida State University, Tallahassee, Florida 32306, USA.
Genome Res. 2023 Dec 1;33(11):2002-2017. doi: 10.1101/gr.277249.122.
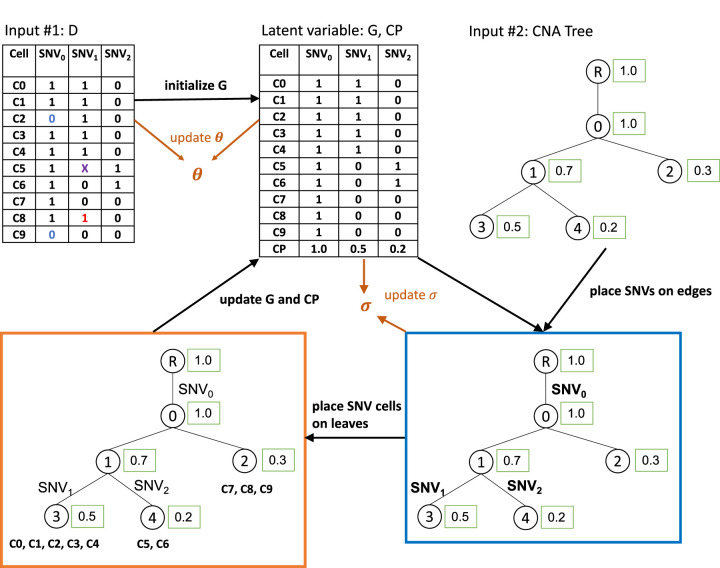
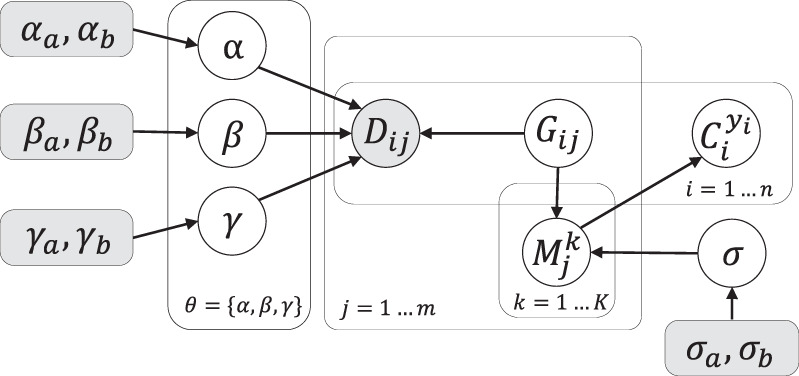
Single-cell DNA sequencing enables the construction of evolutionary trees that can reveal how tumors gain mutations and grow. Different whole-genome amplification procedures render genomic materials of different characteristics, often suitable for the detection of either single-nucleotide variation or copy number aberration, but not ideally for both. Consequently, this hinders the inference of a comprehensive phylogenetic tree and limits opportunities to investigate the interplay of SNVs and CNAs. Existing methods such as SCARLET and COMPASS require that the SNVs and CNAs are detected from the same sets of cells, which is technically challenging. Here we present a novel computational tool, SCsnvcna, that places SNVs on a tree inferred from CNA signals, whereas the sets of cells rendering the SNVs and CNAs are independent, offering a more practical solution in terms of the technical challenges. SCsnvcna is a Bayesian probabilistic model using both the genotype constraints on the tree and the cellular prevalence to search the optimal solution. Comprehensive simulations and comparison with seven state-of-the-art methods show that SCsnvcna is robust and accurate in a variety of circumstances. Particularly, SCsnvcna most frequently produces the lowest error rates, with ability to scale to a wide range of numerical values for leaf nodes in the tree, SNVs, and SNV cells. The application of SCsnvcna to two published colorectal cancer data sets shows highly consistent placement of SNV cells and SNVs with the original study while also supporting a refined placement of ATP7B, illustrating SCsnvcna's value in analyzing complex multitumor samples.
单细胞 DNA 测序能够构建进化树,揭示肿瘤如何获得突变和生长。不同的全基因组扩增程序会产生具有不同特征的基因组材料,通常适用于检测单核苷酸变异或拷贝数异常,但并不理想。因此,这阻碍了对综合系统发育树的推断,并限制了研究 SNV 和 CNA 相互作用的机会。现有的方法,如 SCARLET 和 COMPASS,要求从同一组细胞中检测 SNV 和 CNA,这在技术上具有挑战性。在这里,我们提出了一种新的计算工具 SCsnvcna,它将 SNV 置于从 CNA 信号推断出的树上,而产生 SNV 和 CNA 的细胞集是独立的,在技术挑战方面提供了更实际的解决方案。SCsnvcna 是一种贝叶斯概率模型,同时使用树上的基因型约束和细胞流行率来搜索最优解。全面的模拟和与七种最先进方法的比较表明,SCsnvcna 在各种情况下都具有稳健性和准确性。特别是,SCsnvcna 最常产生最低的错误率,并且能够扩展到树、SNV 和 SNV 细胞的叶节点的广泛数值范围。SCsnvcna 在两个已发表的结直肠癌数据集上的应用表明,SNV 细胞和 SNV 的放置与原始研究高度一致,同时也支持 ATP7B 的精细放置,说明了 SCsnvcna 在分析复杂的多肿瘤样本中的价值。