Epigene Labs, Paris, France.
University of Edinburgh, Edinburgh, UK.
BMC Bioinformatics. 2023 Dec 7;24(1):459. doi: 10.1186/s12859-023-05578-5.
Variability in datasets is not only the product of biological processes: they are also the product of technical biases. ComBat and ComBat-Seq are among the most widely used tools for correcting those technical biases, called batch effects, in, respectively, microarray and RNA-Seq expression data.
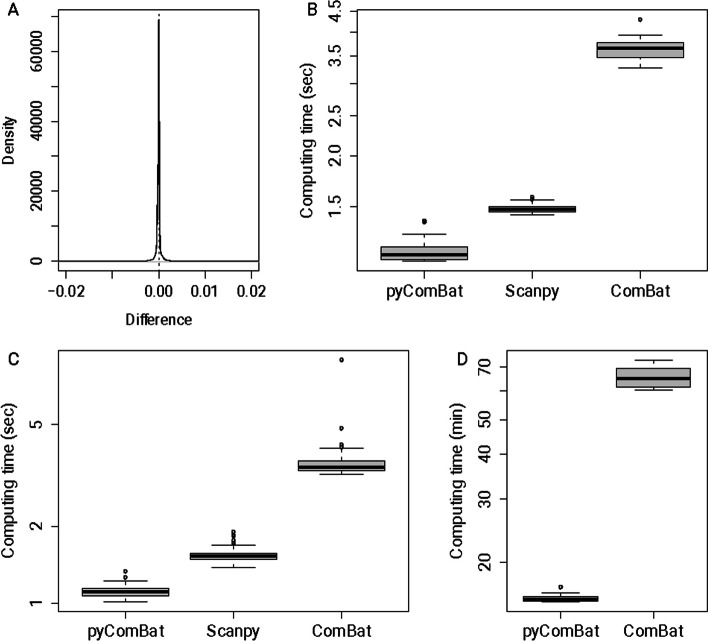
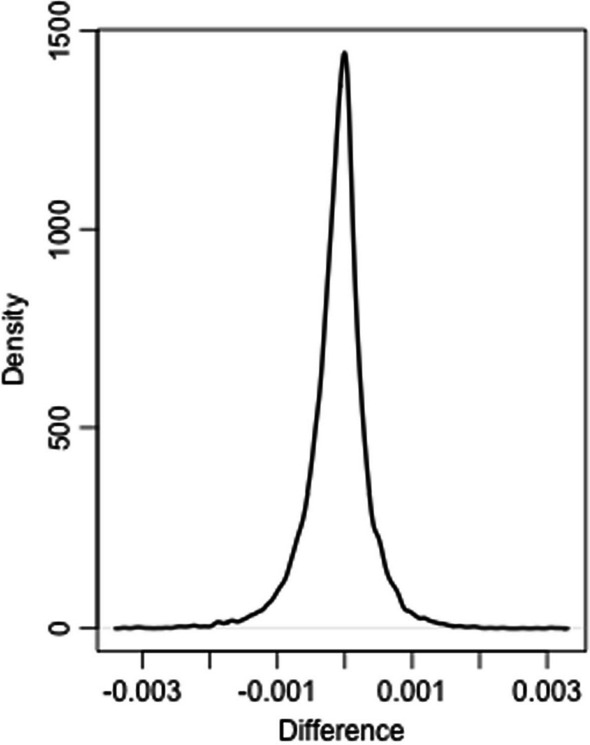
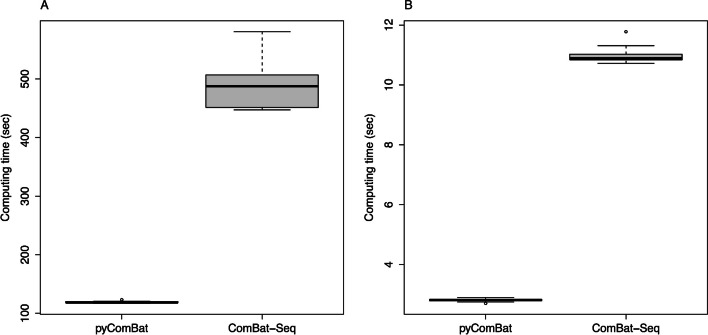
In this technical note, we present a new Python implementation of ComBat and ComBat-Seq. While the mathematical framework is strictly the same, we show here that our implementations: (i) have similar results in terms of batch effects correction; (ii) are as fast or faster than the original implementations in R and; (iii) offer new tools for the bioinformatics community to participate in its development. pyComBat is implemented in the Python language and is distributed under GPL-3.0 ( https://www.gnu.org/licenses/gpl-3.0.en.html ) license as a module of the inmoose package. Source code is available at https://github.com/epigenelabs/inmoose and Python package at https://pypi.org/project/inmoose .
We present a new Python implementation of state-of-the-art tools ComBat and ComBat-Seq for the correction of batch effects in microarray and RNA-Seq data. This new implementation, based on the same mathematical frameworks as ComBat and ComBat-Seq, offers similar power for batch effect correction, at reduced computational cost.
数据集的变异性不仅是生物过程的产物:它们也是技术偏差的产物。ComBat 和 ComBat-Seq 是最广泛用于纠正微阵列和 RNA-Seq 表达数据中所谓批次效应的技术偏差的工具之一。
在本技术说明中,我们提出了 ComBat 和 ComBat-Seq 的新 Python 实现。虽然数学框架完全相同,但我们在这里表明,我们的实现:(i)在批次效应校正方面具有相似的结果;(ii)在速度上与 R 中的原始实现一样快或更快;(iii)为生物信息学社区提供了新的工具来参与其开发。pyComBat 是用 Python 语言实现的,并作为 inmoose 包的一个模块以 GPL-3.0(https://www.gnu.org/licenses/gpl-3.0.en.html)许可证分发。源代码可在 https://github.com/epigenelabs/inmoose 获得,Python 包可在 https://pypi.org/project/inmoose 获得。
我们提出了 ComBat 和 ComBat-Seq 的新的 Python 实现,用于纠正微阵列和 RNA-Seq 数据中的批次效应。这个新的实现基于与 ComBat 和 ComBat-Seq 相同的数学框架,在降低计算成本的同时提供了类似的批次效应校正能力。