An Linna, Said Meerit, Tran Long, Majumder Sagardip, Goreshnik Inna, Lee Gyu Rie, Juergens David, Dauparas Justas, Anishchenko Ivan, Coventry Brian, Bera Asim K, Kang Alex, Levine Paul M, Alvarez Valentina, Pillai Arvind, Norn Christoffer, Feldman David, Zorine Dmitri, Hicks Derrick R, Li Xinting, Sanchez Mariana Garcia, Vafeados Dionne K, Salveson Patrick J, Vorobieva Anastassia A, Baker David
Department of Biochemistry, The University of Washington, Seattle, WA, USA.
Institute for Protein Design, University of Washington, Seattle, WA, USA.
bioRxiv. 2023 Dec 21:2023.12.20.572602. doi: 10.1101/2023.12.20.572602.
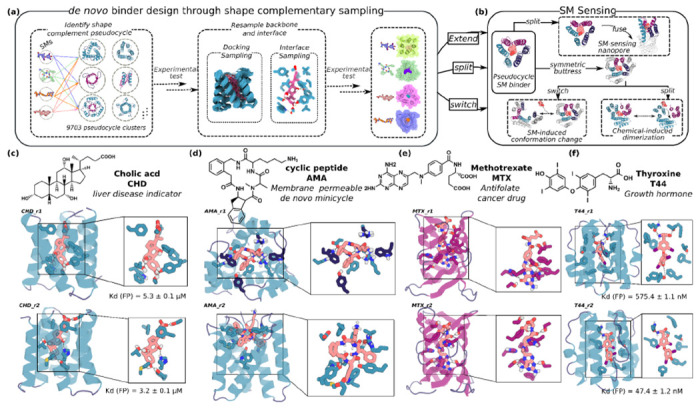
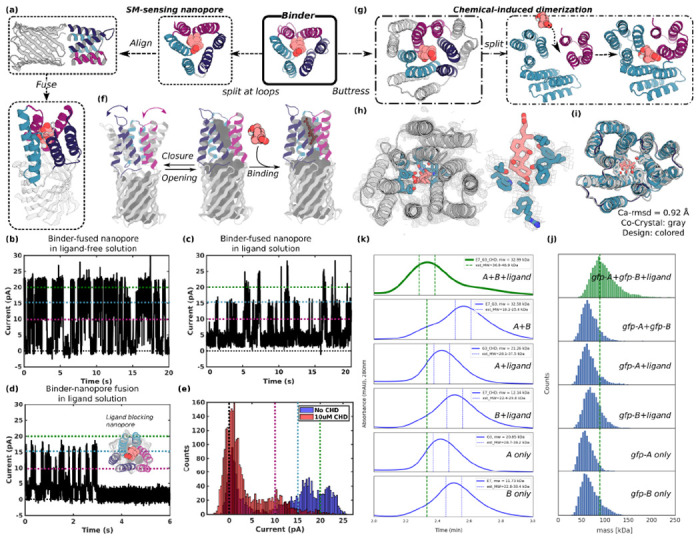
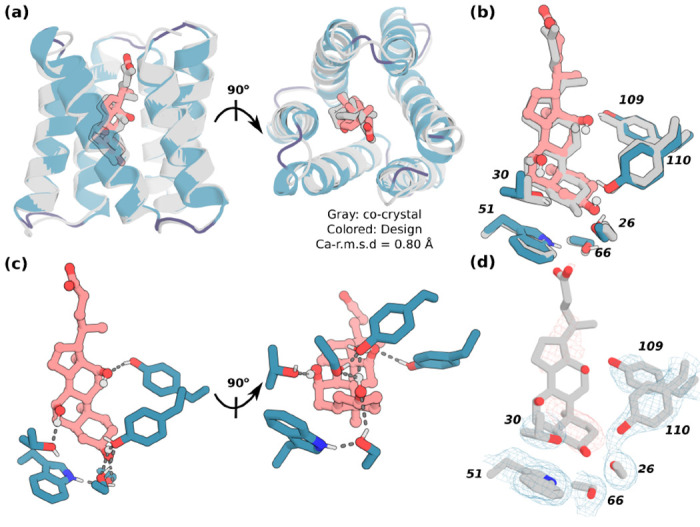
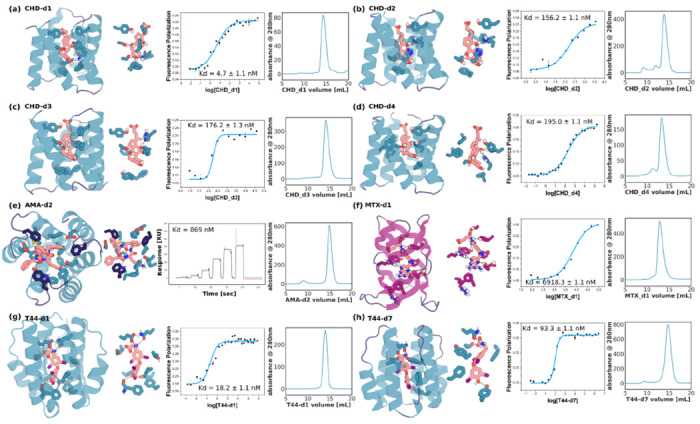
A general method for designing proteins to bind and sense any small molecule of interest would be widely useful. Due to the small number of atoms to interact with, binding to small molecules with high affinity requires highly shape complementary pockets, and transducing binding events into signals is challenging. Here we describe an integrated deep learning and energy based approach for designing high shape complementarity binders to small molecules that are poised for downstream sensing applications. We employ deep learning generated psuedocycles with repeating structural units surrounding central pockets; depending on the geometry of the structural unit and repeat number, these pockets span wide ranges of sizes and shapes. For a small molecule target of interest, we extensively sample high shape complementarity pseudocycles to generate large numbers of customized potential binding pockets; the ligand binding poses and the interacting interfaces are then optimized for high affinity binding. We computationally design binders to four diverse molecules, including for the first time polar flexible molecules such as methotrexate and thyroxine, which are expressed at high levels and have nanomolar affinities straight out of the computer. Co-crystal structures are nearly identical to the design models. Taking advantage of the modular repeating structure of pseudocycles and central location of the binding pockets, we constructed low noise nanopore sensors and chemically induced dimerization systems by splitting the binders into domains which assemble into the original pseudocycle pocket upon target molecule addition.
设计能够结合并感知任何感兴趣小分子的蛋白质的通用方法将具有广泛用途。由于与之相互作用的原子数量较少,要以高亲和力结合小分子需要高度形状互补的口袋,并且将结合事件转化为信号具有挑战性。在此,我们描述了一种基于深度学习和能量的综合方法,用于设计与小分子具有高形状互补性的结合物,这些结合物可用于下游传感应用。我们采用深度学习生成的假环,其具有围绕中央口袋的重复结构单元;根据结构单元的几何形状和重复次数,这些口袋涵盖了广泛的尺寸和形状范围。对于感兴趣的小分子靶标,我们广泛采样高形状互补性假环,以生成大量定制的潜在结合口袋;然后针对高亲和力结合优化配体结合姿势和相互作用界面。我们通过计算设计了针对四种不同分子的结合物,首次包括甲氨蝶呤和甲状腺素等极性柔性分子,它们在计算机设计后能够高水平表达且具有纳摩尔亲和力。共晶体结构与设计模型几乎相同。利用假环的模块化重复结构和结合口袋的中心位置,我们通过将结合物拆分为多个结构域来构建低噪声纳米孔传感器和化学诱导二聚化系统,在加入靶分子后这些结构域会组装成原始的假环口袋。