Liu Lingjie, Zhao Yixin, Siepel Adam
Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
Graduate Program in Genetics, Stony Brook University, Stony Brook, NY.
bioRxiv. 2023 Dec 23:2023.12.21.572932. doi: 10.1101/2023.12.21.572932.
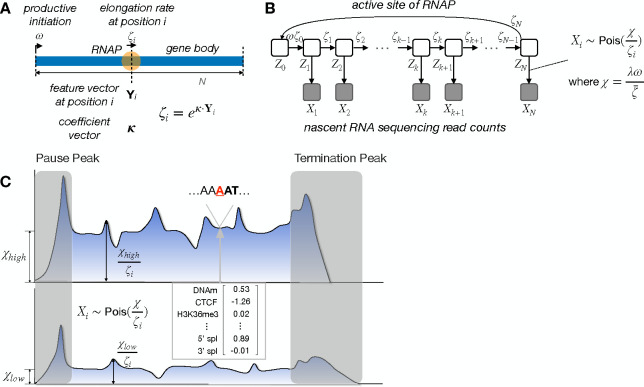
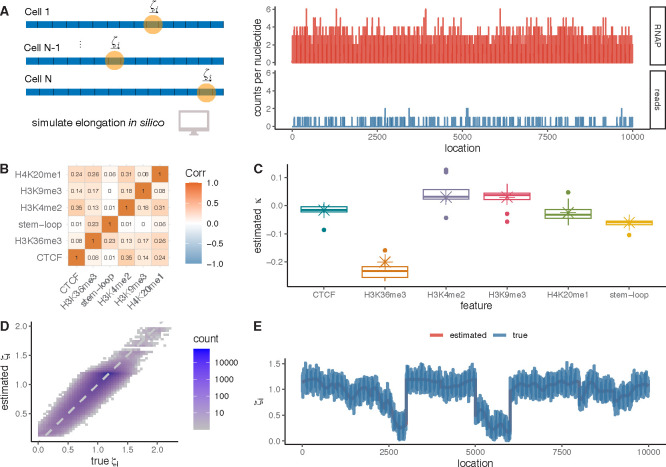
Across all branches of life, transcription elongation is a crucial, regulated phase in gene expression. Many recent studies in eukaryotes have focused on the regulation of promoter-proximal pausing of RNA Polymerase II (Pol II), but rates of productive elongation also vary substantially throughout the gene body, both within and across genes. Here, we introduce a probabilistic model for systematically evaluating potential determinants of the local elongation rate based on nascent RNA sequencing (NRS) data. Our model is derived from a unified model for both the kinetics of Pol II movement along the DNA template and the generation of NRS read counts at steady state. It allows for a continuously variable elongation rate along the gene body, with the rate at each nucleotide defined by a generalized linear relationship with nearby genomic and epigenomic features. High-dimensional feature vectors are accommodated through a sparse-regression extension. We show with simulations that the model allows accurate detection of associated features and accurate prediction of local elongation rates. In an analysis of public PRO-seq and epigenomic data, we identify several features that are strongly associated with reductions in the local elongation rate, including DNA methylation, splice sites, RNA stem-loops, CTCF binding sites, and several histone marks, including H3K36me3 and H4K20me1. By contrast, low-complexity sequences and H3K79me2 marks are associated with increases in elongation rate. In an analysis of DNA -mers, we find that cytosine nucleotides are strongly associated with reductions in local elongation rate, particularly when preceded by guanines and followed by adenines or thymines. Increases in elongation rate are associated with thymines and A+T-rich -mers. These associations are generally shared across cell types, and by considering them our model is effective at predicting features of held-out PRO-seq data. Overall, our analysis is the first to permit genome-wide predictions of relative nucleotide-specific elongation rates based on complex sets of genomic and epigenomic covariates. We have made predictions available for the K562, CD14+, MCF-7, and HeLa-S3 cell types in a UCSC Genome Browser track.
在生命的所有分支中,转录延伸是基因表达中一个关键的、受调控的阶段。最近在真核生物中的许多研究都集中在RNA聚合酶II(Pol II)启动子近端暂停的调控上,但在整个基因体内,无论是在基因内部还是不同基因之间,有效延伸速率也存在很大差异。在这里,我们引入了一个概率模型,用于基于新生RNA测序(NRS)数据系统地评估局部延伸速率的潜在决定因素。我们的模型源自一个统一模型,该模型既描述了Pol II沿DNA模板移动的动力学,又描述了稳态下NRS读数计数的生成。它允许沿基因体的延伸速率连续变化,每个核苷酸处的速率由与附近基因组和表观基因组特征的广义线性关系定义。通过稀疏回归扩展来处理高维特征向量。我们通过模拟表明,该模型能够准确检测相关特征并准确预测局部延伸速率。在对公开的PRO-seq和表观基因组数据的分析中,我们确定了几个与局部延伸速率降低密切相关的特征,包括DNA甲基化、剪接位点、RNA茎环、CTCF结合位点以及几种组蛋白标记,包括H3K36me3和H4K20me1。相比之下,低复杂性序列和H3K79me2标记与延伸速率增加有关。在对DNA -聚体的分析中,我们发现胞嘧啶核苷酸与局部延伸速率降低密切相关,特别是当它前面是鸟嘌呤且后面是腺嘌呤或胸腺嘧啶时。延伸速率增加与胸腺嘧啶和富含A + T的聚体有关。这些关联通常在不同细胞类型中都存在,并且通过考虑这些关联,我们的模型能够有效地预测留存的PRO-seq数据的特征。总体而言,我们的分析首次允许基于复杂的基因组和表观基因组协变量集对全基因组相对核苷酸特异性延伸速率进行预测。我们已在UCSC基因组浏览器轨道中为K562、CD14 +、MCF-7和HeLa-S3细胞类型提供了预测结果。