Dai Jiaqi, Liu Meike, Di Giulio Andrea, Sabatelli Simone, Wang Wenkai, Audisio Paolo
Institute of Entomology, College of Agriculture, Yangtze University, Jingzhou 434025, China.
MARA Key Laboratory of Sustainable Crop Production in the Middle Reaches of the Yangtze River (Co-Construction by Ministry and Province), College of Agriculture, Yangtze University, Jingzhou 434025, China.
Insects. 2024 Jan 12;15(1):57. doi: 10.3390/insects15010057.
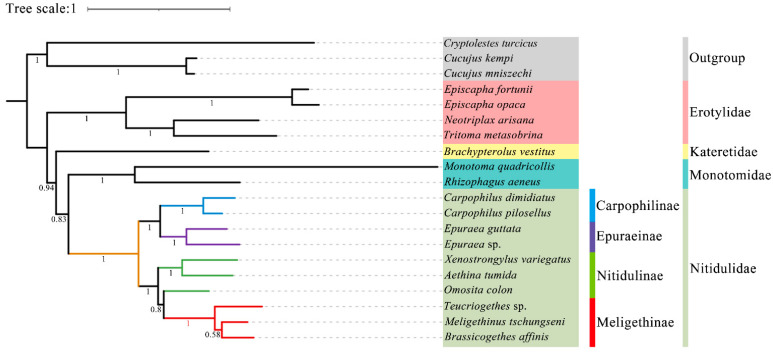
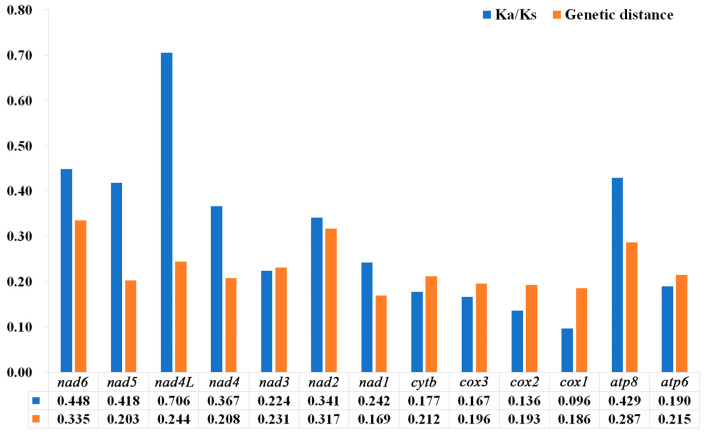
The phylogenetic status of the family Nitidulidae and its sister group relationship remain controversial. Also, the status of the subfamily Meligethinae is not fully understood, and previous studies have been mainly based on morphology, molecular fragments, and biological habits, rather than the analysis of the complete mitochondrial genome. Up to now, there has been no complete mitochondrial genome report of Meligethinae. In this study, the complete mitochondrial genomes of and (both from China) were provided, and they were compared with the existing complete mitochondrial genomes of Nitidulidae. The phylogenetic analysis among 20 species of Coleoptera was reconstructed via PhyloBayes analysis and Maximum likelihood (ML) analysis, respectively. The results showed that the full lengths of and were 15,783 bp and 16,622 bp, and the AT contents were 77% and 76.7%, respectively. Each complete mitochondrial genome contains 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes (rRNAs), and a control region (A + T-rich region). All the PCGs begin with the standard start codon ATN (ATA, ATT, ATG, ATC). All the PCGs terminate with a complete terminal codon, TAA or TAG, except , , , and , which terminate with a single T. Furthermore, all the tRNAs have a typical clover-leaf secondary structure except , whose DHU arm is missing in both species. The two newly sequenced species have different numbers and lengths of tandem repeat regions in their control regions. Based on the genetic distance and Ka/Ks analysis, showed a higher variability and faster evolutionary rate. Based on the available complete mitochondrial genomes, the results showed that the four subfamilies (Nitidulinae, Meligethinae, Carpophilinae, Epuraeinae) of Nitidulidae formed a monophyletic group and further supported the sister group relationship of Nitidulidae + Kateretidae. In addition, the taxonomic status of Meligethinae and the sister group relationship between Meligethinae and Nitidulinae (the latter as currently circumscribed) were also preliminarily explored.
露尾甲科的系统发育地位及其姐妹群关系仍存在争议。此外,扁露尾甲亚科的地位尚未完全明确,以往的研究主要基于形态学、分子片段和生物学习性,而非完整线粒体基因组的分析。截至目前,尚无扁露尾甲亚科完整线粒体基因组的报道。在本研究中,提供了两种(均来自中国)的完整线粒体基因组,并将它们与露尾甲科现有的完整线粒体基因组进行了比较。分别通过贝叶斯系统发育分析和最大似然法(ML)分析重建了20种鞘翅目昆虫的系统发育关系。结果表明,两种的全长分别为15,783 bp和16,622 bp,AT含量分别为77%和76.7%。每个完整的线粒体基因组包含13个蛋白质编码基因(PCGs)、22个转运RNA基因(tRNAs)、2个核糖体RNA基因(rRNAs)和一个控制区(富含A + T的区域)。所有PCGs均以标准起始密码子ATN(ATA、ATT、ATG、ATC)起始。除了 、 、 和 以单个T终止外,所有PCGs均以完整的终止密码子TAA或TAG终止。此外,除了 外,所有tRNAs都具有典型的三叶草二级结构,在这两个物种中其DHU臂均缺失。这两个新测序物种的控制区串联重复区域的数量和长度不同。基于遗传距离和Ka/Ks分析, 表现出更高的变异性和更快的进化速率。基于现有的完整线粒体基因组,结果表明露尾甲科的四个亚科(露尾甲亚科、扁露尾甲亚科、嗜果亚科、埃普露尾甲亚科)形成一个单系群,并进一步支持露尾甲科 + 卡氏甲科的姐妹群关系。此外,还初步探讨了扁露尾甲亚科的分类地位以及扁露尾甲亚科与露尾甲亚科(目前界定的后者)之间的姐妹群关系。