Rahmatpour E, Esmaeili A
Department of Physics, Urmia University, Urmia, Iran.
Sci Rep. 2024 Feb 8;14(1):3226. doi: 10.1038/s41598-024-53341-4.
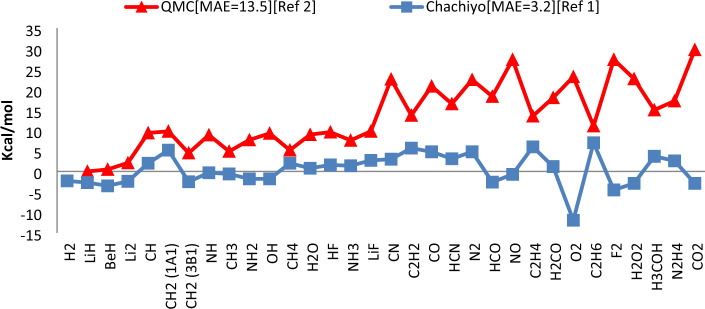
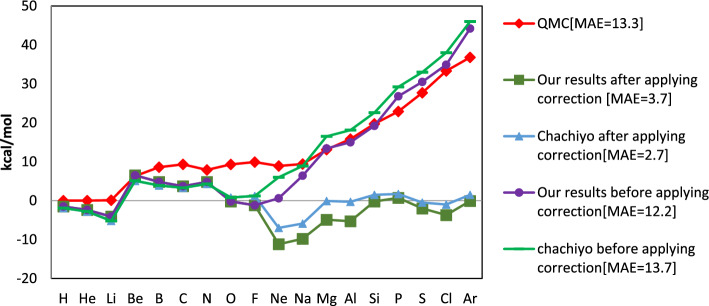
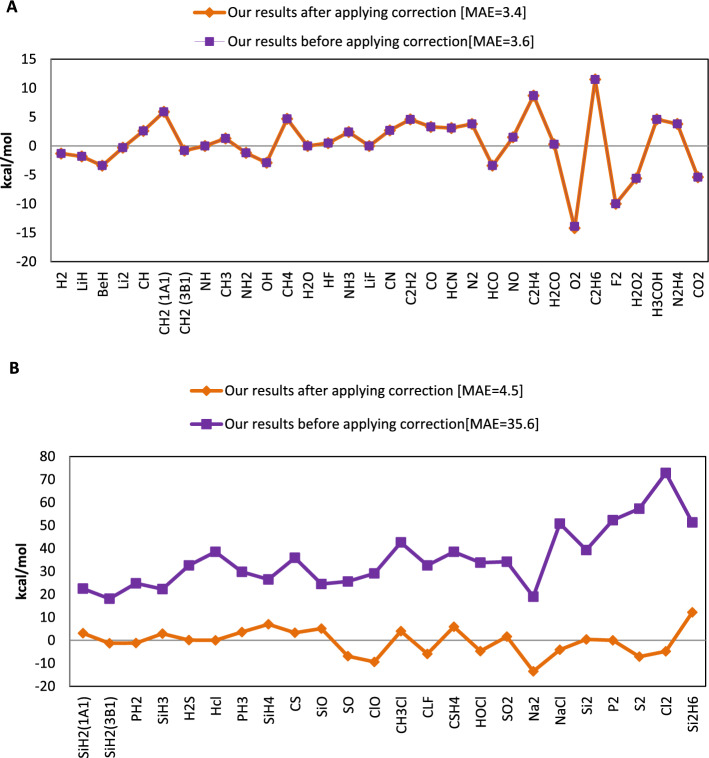
Each of the exchange-correlation functionals in the density functional theory has been customized to particular systems or elements and has unique advantages and disadvantages. In one of the most recent research on exchange-correlation functionals, Chachiyo et al. present a relationship for exchange-correlation functional by assuming the simplest form of electron density. Their utilized electron density causes a systematic inaccuracy in the energy of the molecules since it does not fully account for the variation of the ionization energy for different atoms. We offer a novel relationship for exchange functional that improves the precision of the energy calculations for molecules by using the basic assumptions of the Chachiyo approach and correcting the electron density. Our density is directly related to the atom's ionization energy. Our suggested functional was implemented for 56 molecules composed of atoms from the first, second, and third rows of the periodic table using Siam Quantum package. We discussed about the role of our functional on the reducing the computation error of dipole moment along with total, bonding and zero point energies. We also increased the portion of core electrons to improve the accuracy of the results.
密度泛函理论中的每一种交换关联泛函都是针对特定的体系或元素定制的,都有其独特的优缺点。在最近关于交换关联泛函的一项研究中,Chachiyo等人通过假设最简单形式的电子密度给出了交换关联泛函的一种关系。他们所使用的电子密度导致分子能量出现系统性误差,因为它没有充分考虑不同原子电离能的变化。我们通过采用Chachiyo方法的基本假设并修正电子密度,给出了一种新的交换泛函关系,提高了分子能量计算的精度。我们的密度与原子的电离能直接相关。我们提出的泛函使用暹罗量子软件包,针对由元素周期表第一、二、三周期原子组成的56个分子进行了实现。我们讨论了我们的泛函在降低偶极矩计算误差以及总能量、键能和零点能方面的作用。我们还增加了核心电子的比例以提高结果的准确性。