Rahmatpour Esmaeil, Esmaeili Asghar
Department of Physics, Urmia University, Urmia, Iran.
Sci Rep. 2024 Jul 31;14(1):17715. doi: 10.1038/s41598-024-68655-6.
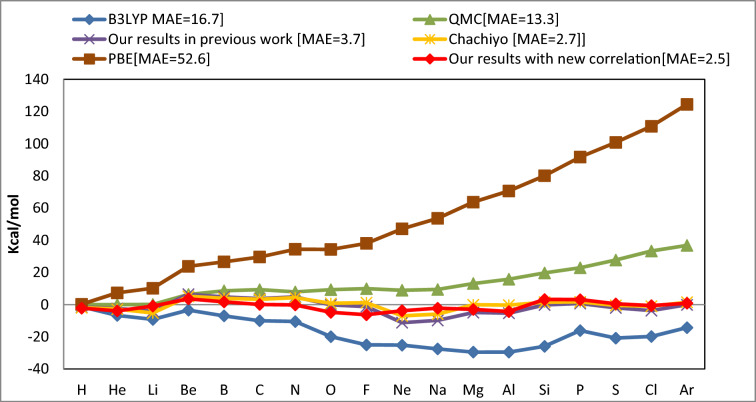
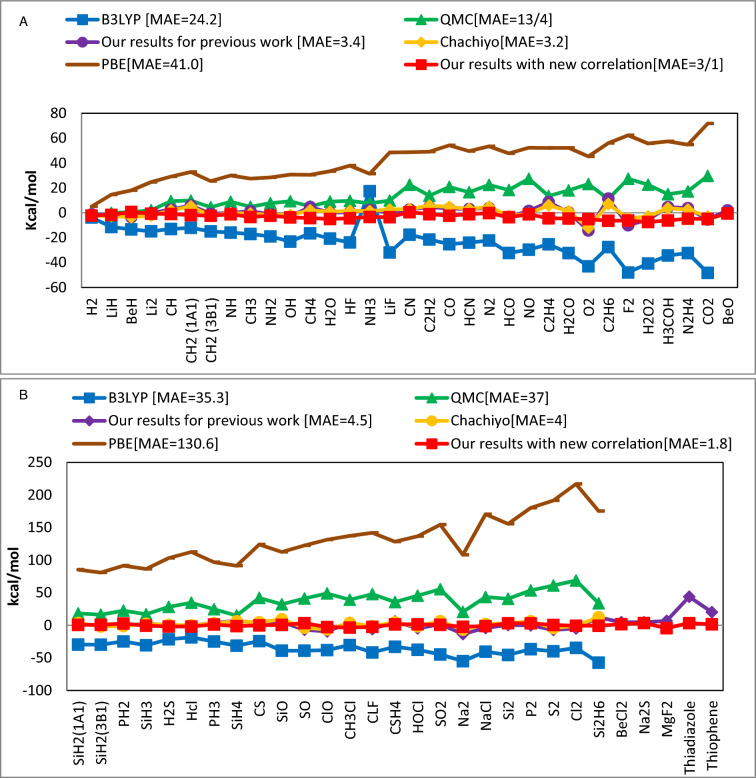
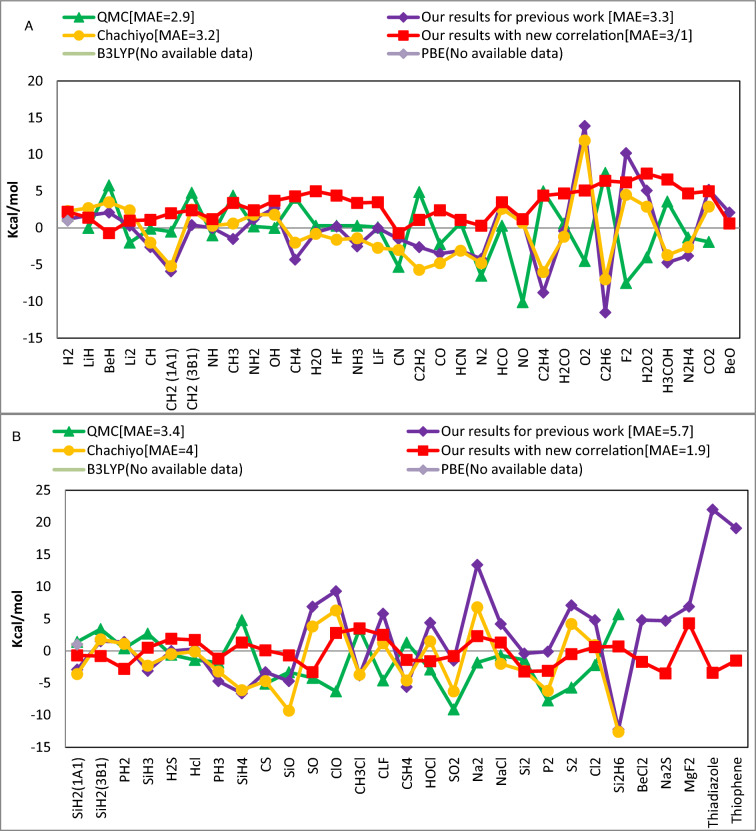
The correlation functional holds significance in density functional theory as it addresses electron-electron interactions beyond the mean-field approximation, enhancing the accuracy of total energy calculations, electronic excitations, and the prediction of materials properties. There are several expressions to describe this energy, and each of them has a unique set of errors in calculating particular properties of materials. This work offers a new correlation functional by employing the density's dependence on ionization energy. We theoretically derived this functional and combined it with the previously reported ionization energy dependent exchange functional to investigate its effect on the total energy, bond energy, dipole moment, and zero-point energy of 62 molecules. The comparison of this new functional in respect to existing widely used correlation models including QMC, PBE, B3LYP and Chachiyo models shows how well it works in producing accurate results with minimal mean absolute error.
关联泛函在密度泛函理论中具有重要意义,因为它处理了超出平均场近似的电子-电子相互作用,提高了总能量计算、电子激发以及材料性质预测的准确性。有几种表达式来描述这种能量,并且它们中的每一个在计算材料的特定性质时都有一组独特的误差。这项工作通过利用密度对电离能的依赖性提供了一种新的关联泛函。我们从理论上推导了这个泛函,并将其与先前报道的依赖于电离能的交换泛函相结合,以研究其对62个分子的总能量、键能、偶极矩和零点能的影响。将这种新泛函与包括QMC、PBE、B3LYP和Chachiyo模型在内的现有广泛使用的关联模型进行比较,显示了它在以最小平均绝对误差产生准确结果方面的效果如何。