Mao Bin, Yang Jie, Zhao Xiaodong, Jia Xueling, Shi Xin, Zhao Lihui, Banerjee Santasree, Zhang Lili, Ma Xiaoling
The Reproductive Medicine Centre, The First Hospital of Lanzhou University, Lanzhou, Gansu 730000, P.R. China.
Department of Genetics, College of Basic Medical Sciences, Jilin University, Changchun, Jilin 130021, P.R. China.
Exp Ther Med. 2024 Jan 11;27(3):97. doi: 10.3892/etm.2024.12385. eCollection 2024 Mar.
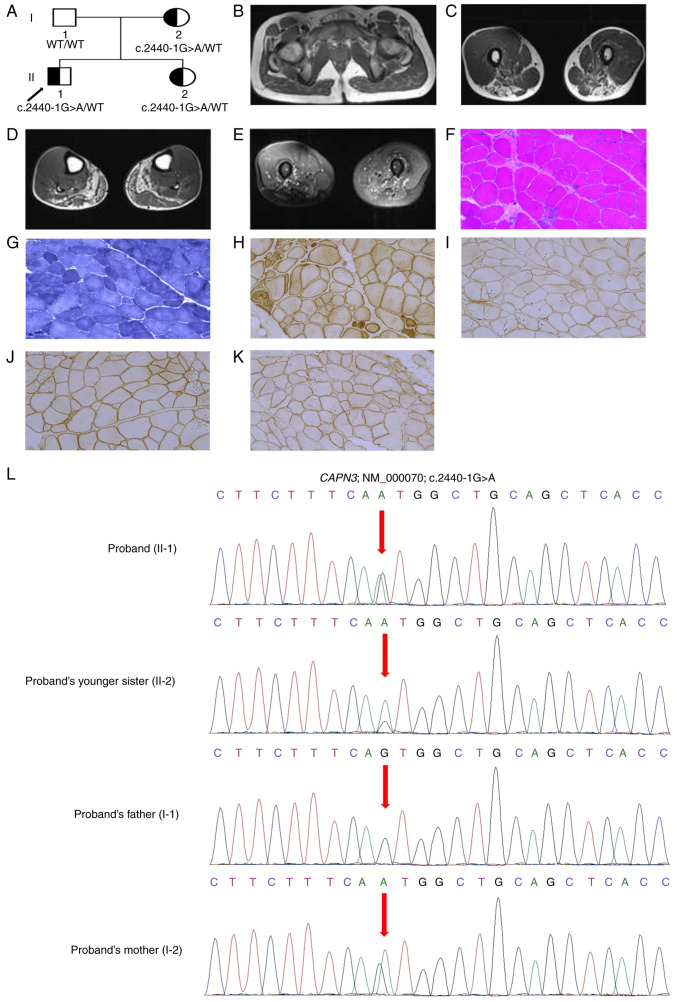
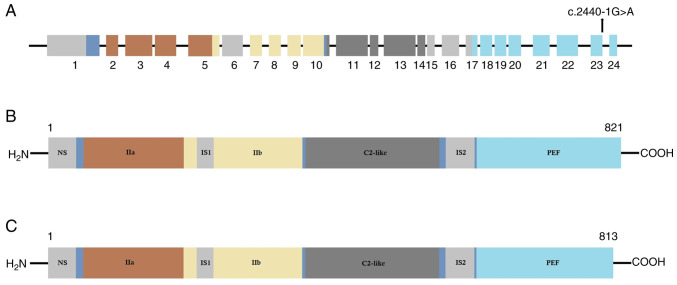
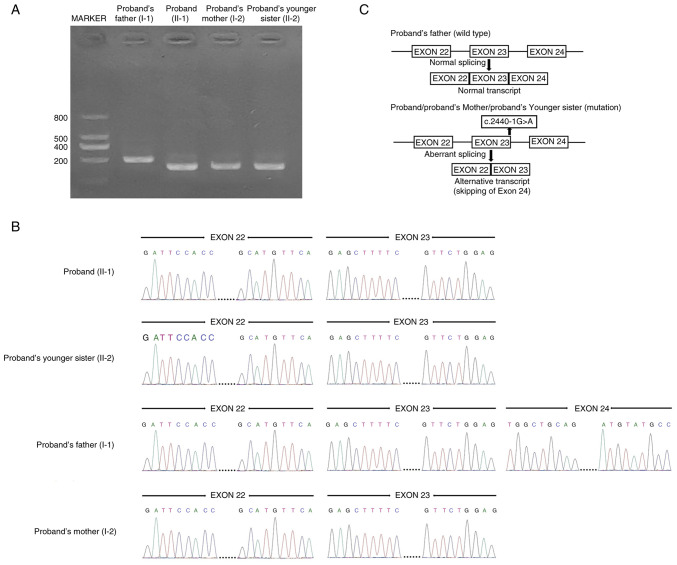
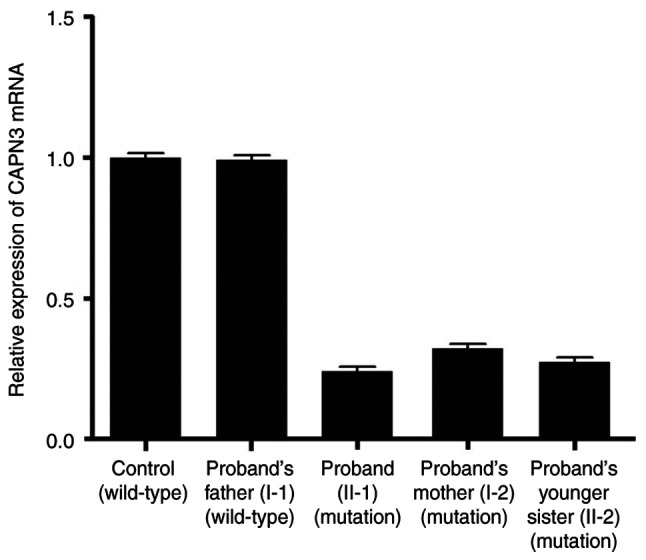
Limb-girdle muscular dystrophies are a group of extremely heterogenous neuromuscular disorders that manifest with gradual and progressive weakness of both proximal and distal muscles. Autosomal dominant limb-girdle muscular dystrophy (LGMDD4) or calpainopathy is a very rare form of myopathy characterized by weakness and atrophy of both proximal and distal muscles with a variable age of onset. LGMDD4 is caused by germline heterozygous mutations of the calpain 3 ( gene. Patients with LGMDD4 often show extreme phenotypic heterogeneity; however, most patients present with gait difficulties, increased levels of serum creatine kinase, myalgia and back pain. In the present study, a 16-year-old male patient, clinically diagnosed with LGMDD4, was investigated. The proband had been suffering from weakness and atrophy of both of their proximal and distal muscles, and had difficulty walking and standing independently. The serum creatine kinase levels (4,754 IU/l; normal, 35-232 IU/l) of the patient were markedly elevated. The younger sister and mother of the proband were also clinically diagnosed with LGMDD4, while the father was phenotypically normal. Whole exome sequencing identified a heterozygous novel splice-site (c.2440-1G>A) mutation in intron 23 of the gene in the proband. Sanger sequencing confirmed that this mutation was also present in both the younger sister and mother of the proband, but the father was not a carrier of this mutation. This splice-site (c.2440-1G>A) mutation causes aberrant splicing of CAPN3 mRNA, leading to the skipping of the last exon (exon 24) of CAPN3 mRNA and resulting in the removal of eight amino acids from the C-terminal of domain IV of the CAPN3 protein. Hence, this splice site mutation causes the formation of a truncated CAPN3 protein (p.Trp814*) of 813 amino acids instead of the wild-type CAPN3 protein that consists of 821 amino acids. This mutation causes partial loss of domain IV (PEF domain) in the CAPN3 protein, which is involved in calcium binding and homodimerization; therefore, this is a loss-of-function mutation. Relative expression of the mutated CAPN3 mRNA was reduced in comparison with the wild-type CAPN3 mRNA in the proband, and their younger sister and mother. This mutation was also not present in 100 normal healthy control individuals of the same ethnicity. The present study reported the first case of gene-associated LGMDD4 in the Chinese population.
肢带型肌营养不良症是一组极其异质性的神经肌肉疾病,表现为近端和远端肌肉逐渐进行性无力。常染色体显性遗传肢带型肌营养不良症(LGMDD4)或钙蛋白酶病是一种非常罕见的肌病形式,其特征是近端和远端肌肉无力和萎缩,发病年龄不一。LGMDD4由钙蛋白酶3(CAPN3)基因的种系杂合突变引起。LGMDD4患者通常表现出极端的表型异质性;然而,大多数患者表现为步态困难、血清肌酸激酶水平升高、肌痛和背痛。在本研究中,对一名临床诊断为LGMDD4的16岁男性患者进行了调查。先证者患有近端和远端肌肉的无力和萎缩,独立行走和站立困难。患者的血清肌酸激酶水平(4754 IU/l;正常范围为35 - 232 IU/l)显著升高。先证者的妹妹和母亲也被临床诊断为LGMDD4,而父亲表型正常。全外显子组测序在先证者的CAPN3基因第23内含子中鉴定出一个杂合的新型剪接位点(c.2440 - 1G>A)突变。桑格测序证实该突变也存在于先证者的妹妹和母亲中,但父亲不是该突变的携带者。这种剪接位点(c.2440 - 1G>A)突变导致CAPN3 mRNA异常剪接,导致CAPN3 mRNA的最后一个外显子(外显子24)缺失,从而导致CAPN3蛋白结构域IV的C末端去除了8个氨基酸。因此,这种剪接位点突变导致形成了一个由813个氨基酸组成的截短的CAPN3蛋白(p.Trp814*),而不是由821个氨基酸组成的野生型CAPN3蛋白。这种突变导致CAPN3蛋白中结构域IV(PEF结构域)部分缺失,该结构域参与钙结合和同源二聚化;因此,这是一个功能丧失突变。与野生型CAPN3 mRNA相比,先证者及其妹妹和母亲中突变的CAPN3 mRNA相对表达量降低。在100名相同种族的正常健康对照个体中也未发现这种突变。本研究报道了中国人群中首例与CAPN3基因相关的LGMDD4病例。