Department of Physiology, Anatomy and Genetics, University of Oxford, Parks Road, Oxford, UK.
Departments of Medicine and Pediatrics, Section of Endocrinology Diabetes and Metabolism, University of Chicago, Chicago, IL, USA.
Diabetologia. 2024 May;67(5):940-951. doi: 10.1007/s00125-024-06103-w. Epub 2024 Feb 17.
AIMS/HYPOTHESIS: The ATP-sensitive potassium (K) channel couples beta cell electrical activity to glucose-stimulated insulin secretion. Loss-of-function mutations in either the pore-forming (inwardly rectifying potassium channel 6.2 [Kir6.2], encoded by KCNJ11) or regulatory (sulfonylurea receptor 1, encoded by ABCC8) subunits result in congenital hyperinsulinism, whereas gain-of-function mutations cause neonatal diabetes. Here, we report a novel loss-of-function mutation (Ser118Leu) in the pore helix of Kir6.2 paradoxically associated with sulfonylurea-sensitive diabetes that presents in early adult life.
A 31-year-old woman was diagnosed with mild hyperglycaemia during an employee screen. After three pregnancies, during which she was diagnosed with gestational diabetes, the patient continued to show elevated blood glucose and was treated with glibenclamide (known as glyburide in the USA and Canada) and metformin. Genetic testing identified a heterozygous mutation (S118L) in the KCNJ11 gene. Neither parent was known to have diabetes. We investigated the functional properties and membrane trafficking of mutant and wild-type K channels in Xenopus oocytes and in HEK-293T cells, using patch-clamp, two-electrode voltage-clamp and surface expression assays.
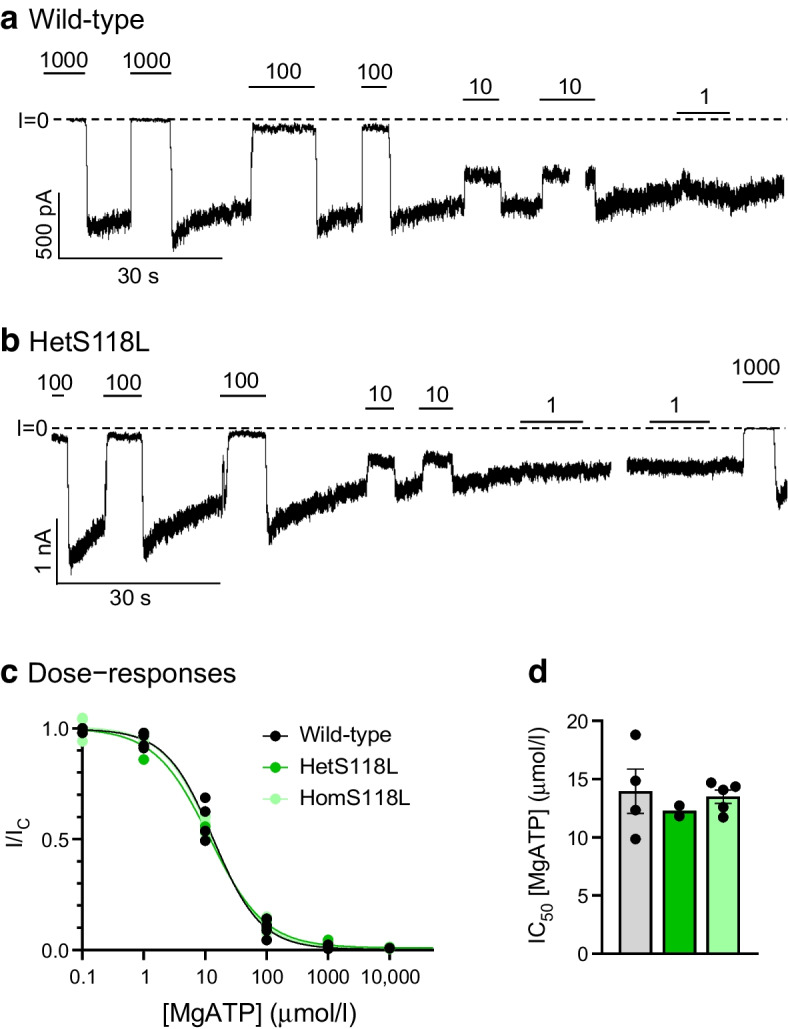
Functional analysis showed no changes in the ATP sensitivity or metabolic regulation of the mutant channel. However, the Kir6.2-S118L mutation impaired surface expression of the K channel by 40%, categorising this as a loss-of-function mutation.
CONCLUSIONS/INTERPRETATION: Our data support the increasing evidence that individuals with mild loss-of-function K channel mutations may develop insulin deficiency in early adulthood and even frank diabetes in middle age. In this case, the patient may have had hyperinsulinism that escaped detection in early life. Our results support the importance of functional analysis of K channel mutations in cases of atypical diabetes.
目的/假设:ATP 敏感性钾 (K) 通道将β细胞电活动与葡萄糖刺激的胰岛素分泌偶联。孔形成(内向整流钾通道 6.2 [Kir6.2],由 KCNJ11 编码)或调节亚基(磺酰脲受体 1,由 ABCC8 编码)的功能丧失突变导致先天性高胰岛素血症,而功能获得性突变导致新生儿糖尿病。在这里,我们报告了一种新的功能丧失突变(Ser118Leu)在 Kir6.2 的孔螺旋中,与磺酰脲敏感的糖尿病相关,该糖尿病在成年早期出现。
一名 31 岁的女性在员工筛查中被诊断为轻度高血糖。在三次妊娠期间,她被诊断为妊娠期糖尿病,此后患者继续出现血糖升高,并接受格列本脲(在美国和加拿大称为glyburide)和二甲双胍治疗。基因检测确定 KCNJ11 基因存在杂合突变(S118L)。父母双方均无糖尿病病史。我们使用膜片钳、双电极电压钳和表面表达测定法,在 Xenopus oocytes 和 HEK-293T 细胞中研究了突变和野生型 K 通道的功能特性和膜转运。
功能分析表明,突变通道的 ATP 敏感性或代谢调节没有变化。然而,Kir6.2-S118L 突变使 K 通道的表面表达减少了 40%,将其归类为功能丧失突变。
结论/解释:我们的数据支持越来越多的证据,即具有轻度功能丧失性 K 通道突变的个体可能在成年早期发展为胰岛素缺乏症,甚至在中年发展为典型糖尿病。在这种情况下,患者可能在早年就有未被发现的高胰岛素血症。我们的结果支持对非典型糖尿病病例中 K 通道突变进行功能分析的重要性。