Department of Civil and Environmental Engineering, Case Western Reserve University, Cleveland, Ohio, USA.
Department of Microbiology, The Ohio State University, Columbus, Ohio, USA.
mSystems. 2024 Mar 19;9(3):e0110523. doi: 10.1128/msystems.01105-23. Epub 2024 Feb 20.
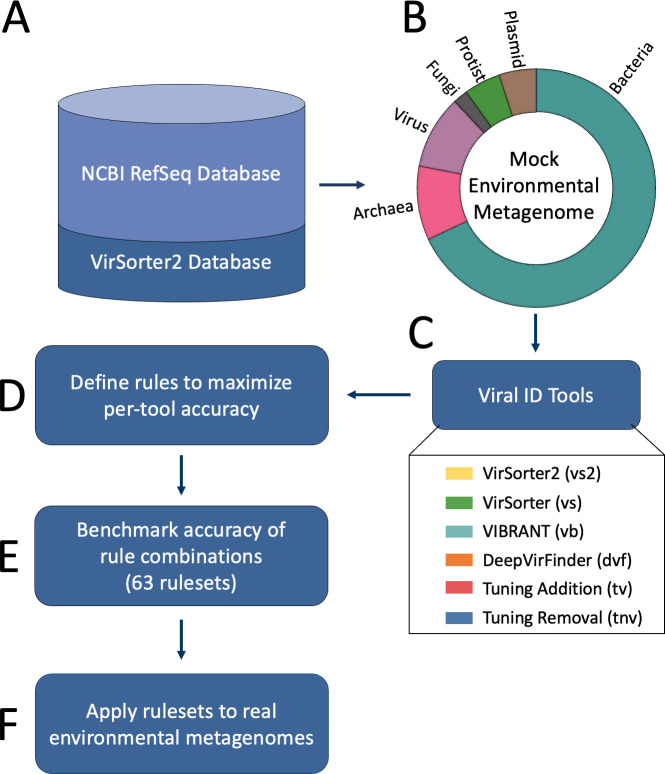
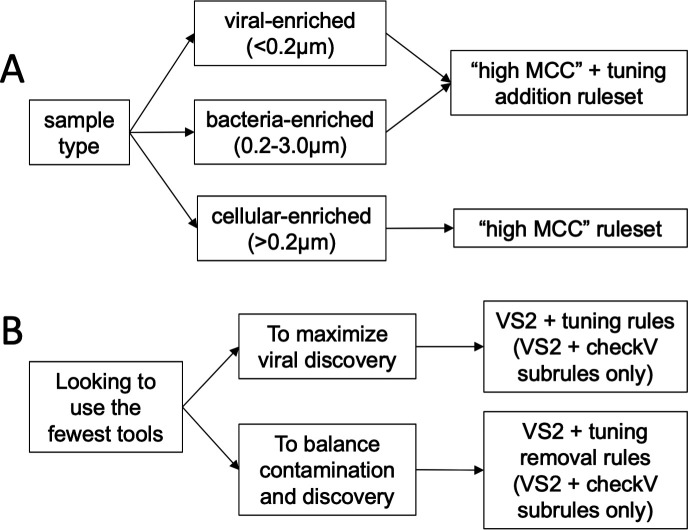
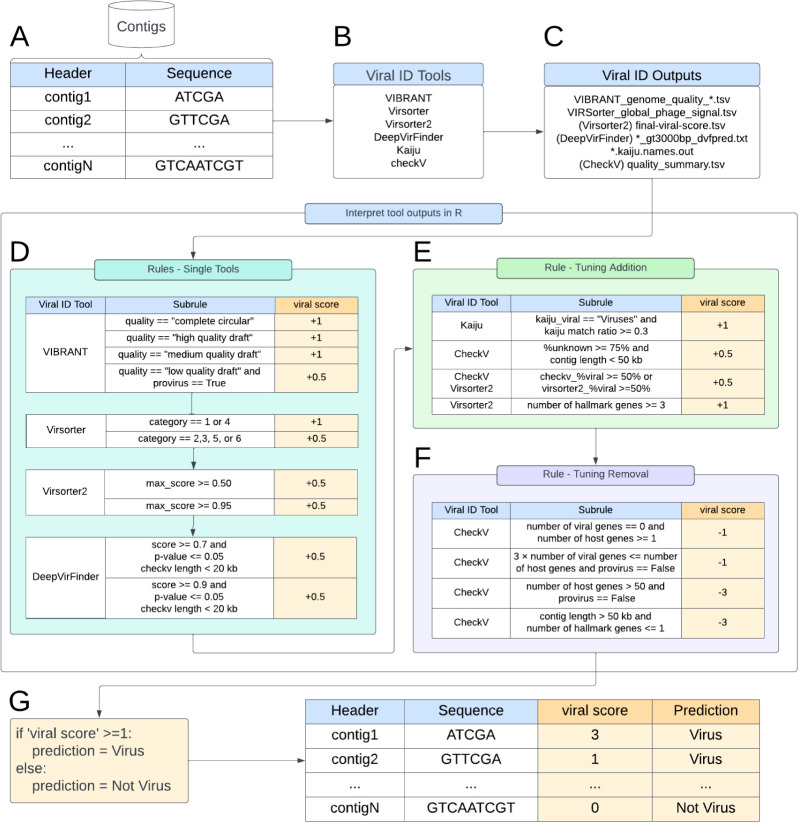
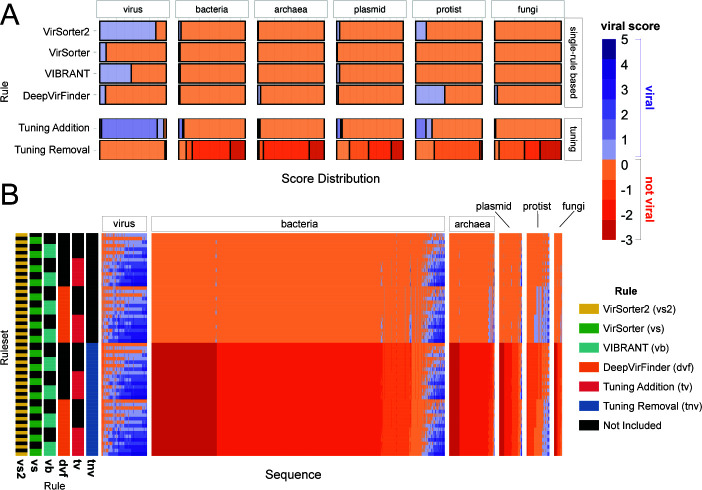
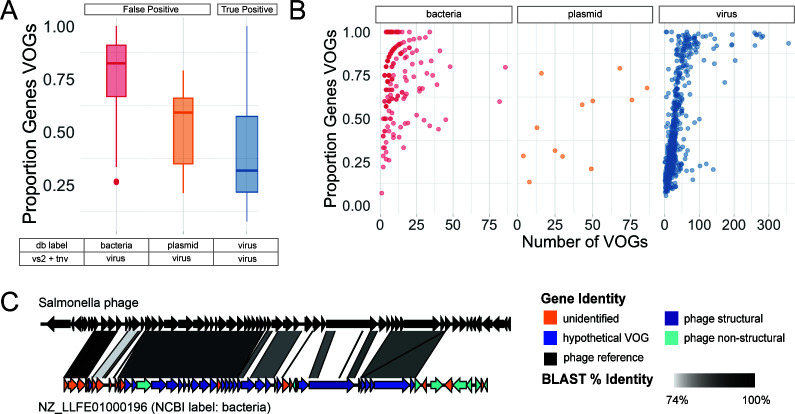
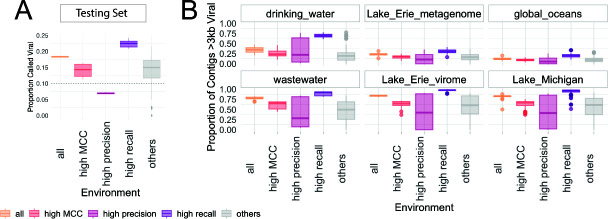
Understanding the ecological impacts of viruses on natural and engineered ecosystems relies on the accurate identification of viral sequences from community sequencing data. To maximize viral recovery from metagenomes, researchers frequently combine viral identification tools. However, the effectiveness of this strategy is unknown. Here, we benchmarked combinations of six widely used informatics tools for viral identification and analysis (VirSorter, VirSorter2, VIBRANT, DeepVirFinder, CheckV, and Kaiju), called "rulesets." Rulesets were tested against mock metagenomes composed of taxonomically diverse sequence types and diverse aquatic metagenomes to assess the effects of the degree of viral enrichment and habitat on tool performance. We found that six rulesets achieved equivalent accuracy [Matthews Correlation Coefficient (MCC) = 0.77, ≥ 0.05]. Each contained VirSorter2, and five used our "tuning removal" rule designed to remove non-viral contamination. While DeepVirFinder, VIBRANT, and VirSorter were each found once in these high-accuracy rulesets, they were not found in combination with each other: combining tools does not lead to optimal performance. Our validation suggests that the MCC plateau at 0.77 is partly caused by inaccurate labeling within reference sequence databases. In aquatic metagenomes, our highest MCC ruleset identified more viral sequences in virus-enriched (44%-46%) than in cellular metagenomes (7%-19%). While improved algorithms may lead to more accurate viral identification tools, this should be done in tandem with careful curation of sequence databases. We recommend using the VirSorter2 ruleset and our empirically derived tuning removal rule. Our analysis provides insight into methods for viral identification and will enable more robust viral identification from metagenomic data sets.
The identification of viruses from environmental metagenomes using informatics tools has offered critical insights in microbial ecology. However, it remains difficult for researchers to know which tools optimize viral recovery for their specific study. In an attempt to recover more viruses, studies are increasingly combining the outputs from multiple tools without validating this approach. After benchmarking combinations of six viral identification tools against mock metagenomes and environmental samples, we found that these tools should only be combined cautiously. Two to four tool combinations maximized viral recovery and minimized non-viral contamination compared with either the single-tool or the five- to six-tool ones. By providing a rigorous overview of the behavior of viral identification strategies and a pipeline to replicate our process, our findings guide the use of existing viral identification tools and offer a blueprint for feature engineering of new tools that will lead to higher-confidence viral discovery in microbiome studies.
理解病毒对自然和工程生态系统的生态影响,依赖于从群落测序数据中准确识别病毒序列。为了从宏基因组中最大限度地回收病毒,研究人员经常结合使用病毒识别工具。然而,这种策略的有效性尚不清楚。在这里,我们对六种广泛使用的病毒识别和分析信息学工具(VirSorter、VirSorter2、VIBRANT、DeepVirFinder、CheckV 和 Kaiju)的组合(称为“规则集”)进行了基准测试。规则集针对由分类多样的序列类型和多样的水生宏基因组组成的模拟宏基因组进行了测试,以评估病毒富集程度和生境对工具性能的影响。我们发现,六个规则集的准确性相当[马修斯相关系数(MCC)=0.77,≥0.05]。每个规则集都包含 VirSorter2,并且五个规则集都使用了我们设计的“调整去除”规则,用于去除非病毒污染。虽然 DeepVirFinder、VIBRANT 和 VirSorter 都在这些高精度规则集中出现过,但它们彼此之间并未组合出现:组合工具并不能带来最佳性能。我们的验证表明,MCC 在 0.77 处达到平台期,部分原因是参考序列数据库中的标签不准确。在水生宏基因组中,我们的最高 MCC 规则集在病毒富集(44%-46%)的样本中比在细胞宏基因组(7%-19%)中识别出更多的病毒序列。虽然改进的算法可能会导致更准确的病毒识别工具,但这应该与序列数据库的仔细管理同时进行。我们建议使用 VirSorter2 规则集和我们经验性推导的调整去除规则。我们的分析提供了对病毒识别方法的深入了解,并将使从宏基因组数据集进行更稳健的病毒识别成为可能。
使用信息学工具从环境宏基因组中鉴定病毒为微生物生态学提供了关键的见解。然而,研究人员仍然难以确定哪些工具最适合他们特定的研究来优化病毒回收。为了尽可能多地回收病毒,研究人员越来越多地结合使用多种工具的输出,而没有验证这种方法。在对六种病毒识别工具的组合与模拟宏基因组和环境样本进行基准测试后,我们发现这些工具的组合应该谨慎进行。与单个工具或五到六个工具相比,两到四个工具的组合最大限度地提高了病毒的回收,同时最大限度地减少了非病毒污染。通过对病毒识别策略的行为进行严格概述,并提供一个可复制我们流程的管道,我们的研究结果为现有病毒识别工具的使用提供了指导,并为新工具的特征工程提供了蓝图,这将有助于在微生物组研究中更有信心地发现病毒。