Glickman Cody, Hendrix Jo, Strong Michael
Center for Genes, Environment, and Health, National Jewish Health, 1400 Jackson Street, Denver, CO, 80206, USA.
Computational Bioscience, University of Colorado Anschutz, 12801 E 17th Avenue, Aurora, CO, 80045, USA.
BMC Bioinformatics. 2021 Jun 16;22(1):329. doi: 10.1186/s12859-021-04242-0.
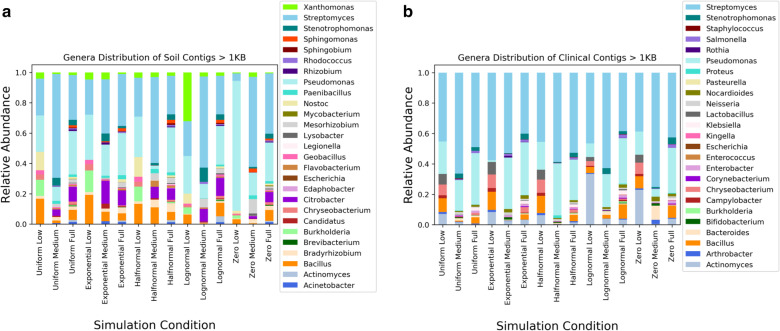
Viruses, including bacteriophages, are important components of environmental and human associated microbial communities. Viruses can act as extracellular reservoirs of bacterial genes, can mediate microbiome dynamics, and can influence the virulence of clinical pathogens. Various targeted metagenomic analysis techniques detect viral sequences, but these methods often exclude large and genome integrated viruses. In this study, we evaluate and compare the ability of nine state-of-the-art bioinformatic tools, including Vibrant, VirSorter, VirSorter2, VirFinder, DeepVirFinder, MetaPhinder, Kraken 2, Phybrid, and a BLAST search using identified proteins from the Earth Virome Pipeline to identify viral contiguous sequences (contigs) across simulated metagenomes with different read distributions, taxonomic compositions, and complexities.
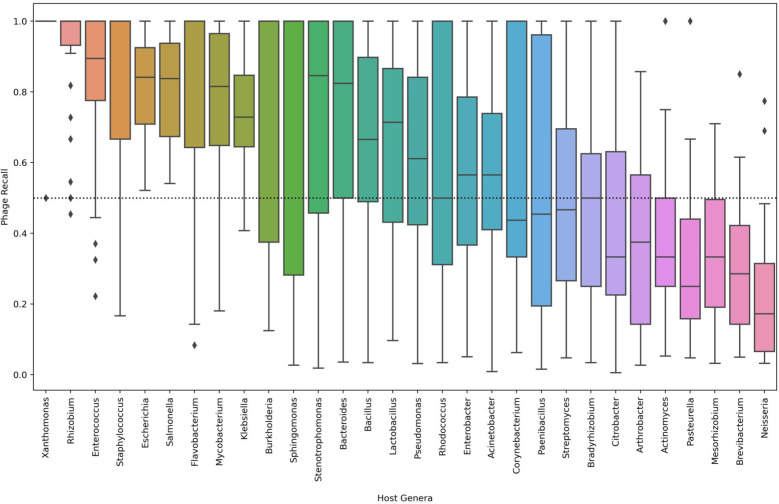
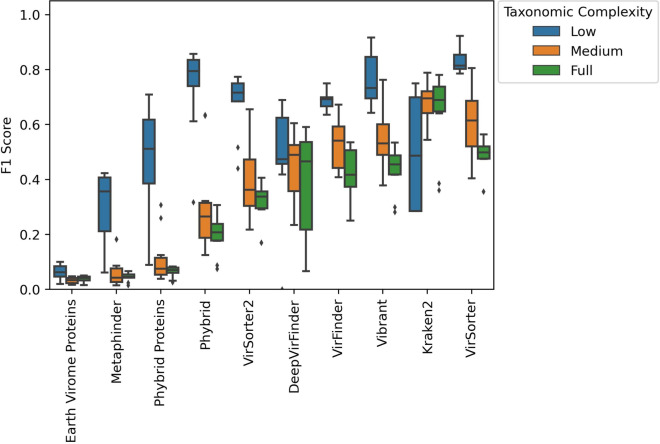
Of the tools tested in this study, VirSorter achieved the best F1 score while Vibrant had the highest average F1 score at predicting integrated prophages. Though less balanced in its precision and recall, Kraken2 had the highest average precision by a substantial margin. We introduced the machine learning tool, Phybrid, which demonstrated an improvement in average F1 score over tools such as MetaPhinder. The tool utilizes machine learning with both gene content and nucleotide features. The addition of nucleotide features improves the precision and recall compared to the gene content features alone.Viral identification by all tools was not impacted by underlying read distribution but did improve with contig length. Tool performance was inversely related to taxonomic complexity and varied by the phage host. For instance, Rhizobium and Enterococcus phages were identified consistently by the tools; whereas, Neisseria prophage sequences were commonly missed in this study.
This study benchmarked the performance of nine state-of-the-art bioinformatic tools to identify viral contigs across different simulation conditions. This study explored the ability of the tools to identify integrated prophage elements traditionally excluded from targeted sequencing approaches. Our comprehensive analysis of viral identification tools to assess their performance in a variety of situations provides valuable insights to viral researchers looking to mine viral elements from publicly available metagenomic data.
病毒,包括噬菌体,是环境微生物群落和人类相关微生物群落的重要组成部分。病毒可作为细菌基因的胞外储存库,介导微生物群落动态变化,并可影响临床病原体的毒力。各种靶向宏基因组分析技术可检测病毒序列,但这些方法通常会排除大型病毒和基因组整合病毒。在本研究中,我们评估并比较了九种最先进的生物信息学工具的能力,这些工具包括Vibrant、VirSorter、VirSorter2、VirFinder、DeepVirFinder、MetaPhinder、Kraken 2、Phybrid,以及使用来自地球病毒组管道中已鉴定蛋白质的BLAST搜索,以识别跨越具有不同读段分布、分类组成和复杂度的模拟宏基因组的病毒连续序列(重叠群)。
在本研究测试的工具中,VirSorter在预测整合原噬菌体时获得了最佳F1分数,而Vibrant在预测整合原噬菌体方面具有最高的平均F1分数。尽管Kraken2的精确率和召回率不太平衡,但其平均精确率却大幅领先。我们引入了机器学习工具Phybrid,它在平均F1分数上比MetaPhinder等工具有所提高。该工具利用机器学习结合基因内容和核苷酸特征。与仅使用基因内容特征相比,添加核苷酸特征提高了精确率和召回率。所有工具的病毒鉴定均不受潜在读段分布的影响,但会随着重叠群长度的增加而提高。工具性能与分类复杂度呈负相关,且因噬菌体宿主而异。例如,工具能够一致地鉴定出根瘤菌噬菌体和肠球菌噬菌体;而在本研究中,淋病奈瑟菌原噬菌体序列常常被遗漏。
本研究对九种最先进的生物信息学工具在不同模拟条件下识别病毒重叠群的性能进行了基准测试。本研究探索了这些工具识别传统上被靶向测序方法排除的整合原噬菌体元件的能力。我们对病毒鉴定工具在各种情况下的性能进行的全面分析,为希望从公开可用的宏基因组数据中挖掘病毒元件的病毒研究人员提供了有价值的见解。